 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Pathophysiology of Heart Failure
Cardiac dysfunction precipitates changes in vascular function, blood volume, and neurohumoral status. These changes serve as compensatory mechanisms to help maintain cardiac output (primarily by the Frank-Starling mechanism) and arterial blood pressure (by systemic vasoconstriction). However, these compensatory changes over months and years can worsen cardiac function. Therefore, some of the most effective treatments for managing chronic heart failure involve modulating non-cardiac factors such as arterial and venous pressures by administering vasodilator and diuretic drugs.
Cardiac function
 Changes in cardiac function associated with heart failure decrease cardiac output. This results from a decline in stroke volume that is due to systolic dysfunction, diastolic dysfunction, or a combination of the two. Briefly, systolic dysfunction results from a loss of intrinsic inotropy (contractility), which can be caused by disease-induced alterations in signal transduction mechanisms responsible for regulating inotropy. It can also result from acute or chronic ischemia, or from the loss of viable, contracting muscle following acute myocardial infarction. Diastolic dysfunction refers to the diastolic properties of the ventricle and occurs when the ventricle becomes less compliant (i.e., "stiffer"), which impairs ventricular filling. Reduced filling of the ventricle results in less ejection of blood. Both systolic and diastolic dysfunction lead to an increase in ventricular end-diastolic pressure, which serves as a compensatory mechanism through utilization of the Frank-Starling mechanism to augment stroke volume. In some types of heart failure (e.g., dilated cardiomyopathy), the ventricle dilates anatomically, which helps to partially normalize the elevated preload pressures by accommodating the increase in end-diastolic volume.
Changes in cardiac function associated with heart failure decrease cardiac output. This results from a decline in stroke volume that is due to systolic dysfunction, diastolic dysfunction, or a combination of the two. Briefly, systolic dysfunction results from a loss of intrinsic inotropy (contractility), which can be caused by disease-induced alterations in signal transduction mechanisms responsible for regulating inotropy. It can also result from acute or chronic ischemia, or from the loss of viable, contracting muscle following acute myocardial infarction. Diastolic dysfunction refers to the diastolic properties of the ventricle and occurs when the ventricle becomes less compliant (i.e., "stiffer"), which impairs ventricular filling. Reduced filling of the ventricle results in less ejection of blood. Both systolic and diastolic dysfunction lead to an increase in ventricular end-diastolic pressure, which serves as a compensatory mechanism through utilization of the Frank-Starling mechanism to augment stroke volume. In some types of heart failure (e.g., dilated cardiomyopathy), the ventricle dilates anatomically, which helps to partially normalize the elevated preload pressures by accommodating the increase in end-diastolic volume.
Therapeutic interventions to improve cardiac function in acute heart failure include the use of cardiostimulatory drugs that stimulate heart rate and contractility, and vasodilator drugs that reduce ventricular afterload and thereby enhance stroke volume. Vasodilator drugs are important in the management of chronic heart failure; cardiostimulatory drugs are only used in acute heart failure or in chronic heart failure patients that undergo acute cardiac decompensation.
Neurohumoral Changes
 Neurohumoral responses occur during heart failure. These include activation of sympathetic nerves and the renin-angiotensin system, and increased release of antidiuretic hormone (vasopressin) and atrial natriuretic peptide. The net effect of these neurohumoral responses is to produce arterial vasoconstriction (to help maintain arterial pressure), venous constriction (to increase venous pressure), and increased blood volume to increase ventricular filling. Neurohumoral activation is an important compensatory response, but it can also aggravate heart failure by increasing ventricular afterload (which depresses stroke volume) and increasing preload to the point where pulmonary or systemic congestion and edema occur. Therefore, it is essential to understand the pathophysiology of heart failure because it serves as the rationale for therapeutic intervention.
Neurohumoral responses occur during heart failure. These include activation of sympathetic nerves and the renin-angiotensin system, and increased release of antidiuretic hormone (vasopressin) and atrial natriuretic peptide. The net effect of these neurohumoral responses is to produce arterial vasoconstriction (to help maintain arterial pressure), venous constriction (to increase venous pressure), and increased blood volume to increase ventricular filling. Neurohumoral activation is an important compensatory response, but it can also aggravate heart failure by increasing ventricular afterload (which depresses stroke volume) and increasing preload to the point where pulmonary or systemic congestion and edema occur. Therefore, it is essential to understand the pathophysiology of heart failure because it serves as the rationale for therapeutic intervention.
There is also evidence that other factors such as nitric oxide and endothelin (both of which are increased in heart failure) may play a role in the pathogenesis of heart failure.
Some drug treatments for heart failure involve attenuating the neurohumoral changes. For example, certain beta-blockers have been shown to provide significant long-term benefit, quite likely because they block the effects of excessive sympathetic activation on the heart. Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and aldosterone receptor antagonists are commonly used to treat heart failure by inhibiting the actions of the renin-angiotensin-aldosterone system.
Systemic Vascular Function
To compensate for reduced cardiac output during heart failure, feedback mechanisms within the body try to maintain normal arterial pressure. This occurs by constricting arterial resistance vessels (increasing systemic vascular resistance) through activation of the sympathetic adrenergic nervous system, thereby increasing systemic vascular resistance. Veins are also constricted to elevate venous pressure. Arterial baroreceptors are important components of this feedback system, especially in acute heart failure. Activation of the renin-angiotensin system and antidiuretic hormone (vasopressin) also contribute to systemic vasoconstriction.
Drugs that block some of these neurohumoral mechanisms, such angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, improve ventricular stroke volume by reducing afterload on the ventricle. Vasodilator drugs such as hydralazine and sodium nitroprusside are also used to reduce afterload on the ventricle and thereby enhance cardiac output.
Blood Volume
In heart failure, there is a compensatory increase in blood volume that serves to increase ventricular preload and thereby enhance stroke volume by the Frank-Starling mechanism. Blood volume is augmented by several factors. Reduced renal perfusion results in decreased urine output and retention of fluid. Furthermore, a combination of reduced renal perfusion and sympathetic activation of the kidneys stimulates the release of renin, thereby activating the renin-angiotensin system and enhancing aldosterone secretion. There is also an increase in circulating arginine vasopressin (antidiuretic hormone) that contributes to renal retention of water. The result of humoral activation is an increase in renal reabsorption of sodium and water, which increases blood volume to help maintain cardiac output; however, the increased volume can be deleterious because it raises venous pressures, which can lead to pulmonary and systemic edema. Pulmonary edema causes exertional dyspnea (shortness of breath during exertion). Therefore, most patients in heart failure are treated with diuretic drugs to reduce blood volume and venous pressures to reduce edema.
Integration of Cardiac and Vascular Changes
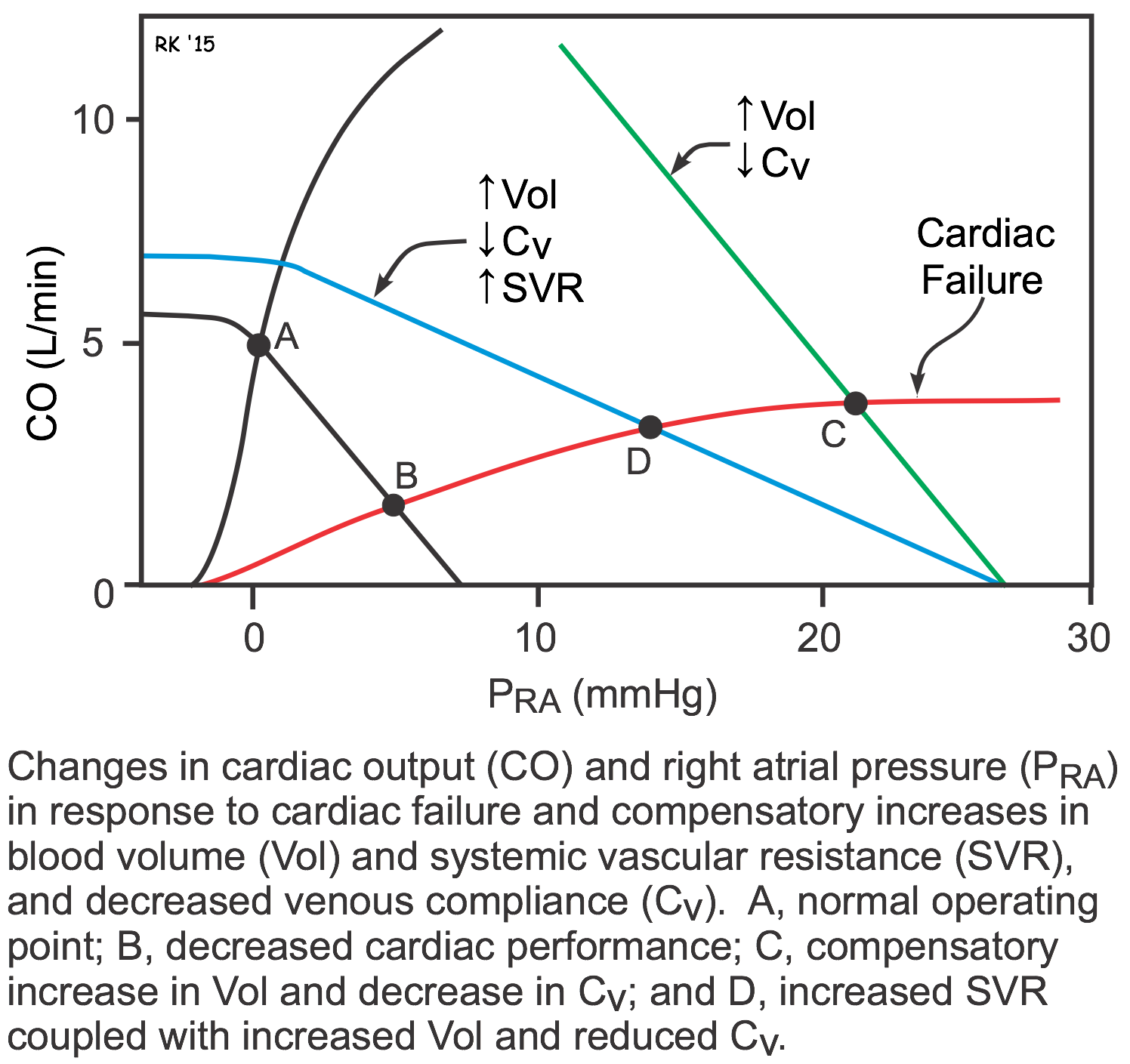 As described above, both systolic and diastolic heart failure lead to changes in systemic vascular resistance, blood volume, and venous pressures. These changes can be examined graphically by using cardiac and vascular function curves, as shown to the right. The decrease in cardiac performance causes a downward shift in the slope of the cardiac function curve. This alone would lead to an increase in right atrial or central venous pressure (point B) as well as a large decrease in cardiac output. The increase in blood volume and venoconstriction (decreased venous compliance) causes a parallel shift to the right of the systemic vascular function curve (green line; point C). Because systemic vascular resistance also increases, the slope of the vascular function curve shifts downward (blue line; point D). These changes in vascular function, coupled with the downward shift in the cardiac function curve, cause a large increase in right atrial or central venous pressure (point D). This partially helps to offset the large decline in cardiac output that would occur in the absence of the systemic vascular responses (point B). Therefore, the systemic responses (vascular constriction and increased blood volume) help to compensate for the loss of cardiac performance; however, these compensatory responses cause a large increase in venous pressure that can lead to edema. Furthermore, the increase in systemic vascular resistance increases the afterload on the left ventricle, which can further depress its output.
As described above, both systolic and diastolic heart failure lead to changes in systemic vascular resistance, blood volume, and venous pressures. These changes can be examined graphically by using cardiac and vascular function curves, as shown to the right. The decrease in cardiac performance causes a downward shift in the slope of the cardiac function curve. This alone would lead to an increase in right atrial or central venous pressure (point B) as well as a large decrease in cardiac output. The increase in blood volume and venoconstriction (decreased venous compliance) causes a parallel shift to the right of the systemic vascular function curve (green line; point C). Because systemic vascular resistance also increases, the slope of the vascular function curve shifts downward (blue line; point D). These changes in vascular function, coupled with the downward shift in the cardiac function curve, cause a large increase in right atrial or central venous pressure (point D). This partially helps to offset the large decline in cardiac output that would occur in the absence of the systemic vascular responses (point B). Therefore, the systemic responses (vascular constriction and increased blood volume) help to compensate for the loss of cardiac performance; however, these compensatory responses cause a large increase in venous pressure that can lead to edema. Furthermore, the increase in systemic vascular resistance increases the afterload on the left ventricle, which can further depress its output.
Revised 11/05/2023