 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Cardiac Excitation-Contraction Coupling
Excitation-contraction coupling (ECC) is the process whereby an action potential triggers a myocyte to contract, followed by subsequent relaxation. The following figure and text summarize some of the key events that occur during cardiac muscle excitation-contraction coupling:
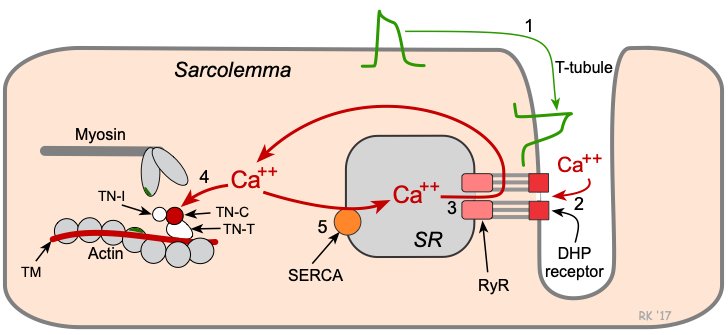
Key steps in cardiac excitation-contraction coupling:
- Action potentials traveling along the sarcolemma and down into the transverse tubule (T-tubule) system depolarize the cell membrane.
- Voltage-sensitive dihydropyridine (DHP) receptors (L-type calcium channels) open, which permits calcium entry into the cell during phase 2 of the action potential.
- Calcium influx triggers a subsequent release of calcium that is stored in the sarcoplasmic reticulum (SR) through calcium-release channels ("ryanodine receptors"), and increases intracellular calcium concentration from about 10-7 to 10-5 M.
- Free calcium binds to troponin-C (TN-C) that is part of the regulatory complex attached to the thin filaments. When calcium binds to the TN-C, this induces a conformational change in the regulatory complex such that troponin-I (TN-I) exposes a site on the actin molecule that can bind to the myosin ATPase on the myosin head. This binding results in ATP hydrolysis that supplies energy for a conformational change to occur in the actin-myosin complex. The result is a movement ("ratcheting") between the myosin heads and the actin, such that the actin and myosin filaments slide past each other, shortening the sarcomere length. Ratcheting cycles occur as long as the cytosolic calcium remains elevated.
- At the end of phase 2, calcium entry into the cell slows and calcium is sequestered by the SR by an ATP-dependent calcium pump (SERCA, sarco-endoplasmic reticulum calcium-ATPase), thus lowering the cytosolic calcium concentration and removing calcium from the TN-C. To a quantitatively smaller extent, cytosolic calcium is transported out of the cell by the sodium-calcium-exchange pump. Unbinding of calcium from TN-C induces a conformational change in the troponin complex, leading, once again, to TN-I inhibition of the actin binding site. At the end of the cycle, a new ATP binds to the myosin head, displacing the ADP, and the initial sarcomere length is restored.
Mechanisms that enhance the concentration of cytosolic calcium increase the amount of ATP hydrolyzed, and increase the force generated by the actin and myosin interactions, as well as the velocity of shortening. Physiologically, cytosolic calcium concentrations are influenced primarily by beta-adrenoceptor-coupled mechanisms. Beta-adrenergic stimulation, as occurs when sympathetic nerves are activated, increases cAMP, which activates protein kinase to increase in calcium entry into the cell through L-type calcium channels. Activation of the IP3 signal transduction pathway also can stimulate the release of calcium by the SR through IP3 receptors on the SR. Activation of the cAMP-dependent protein kinase phosphorylates a protein (phospholamban) on the SR that normally inhibits calcium uptake. This disinhibition of phospholamban increases the rate of calcium uptake by the SR. Therefore, beta-adrenergic stimulation increases the force and shortening velocity of contraction (i.e., positive inotropy), and increases the rate of relaxation (i.e., positive lusitropy).
Another potential regulatory mechanism for ECC involves altering the affinity of TN-C for calcium. There are investigational drugs that enhance TN-C calcium affinity and exert a positive inotropic influence on the heart. One potential downside to these drugs, however, is that enhanced TN-C binding to calcium can reduce the rate of relaxation, causing diastolic dysfunction.
In systolic heart failure, ECC can be impaired at several sites. First, there can be a decreased influx of calcium into the cell through L-type calcium channels (resulting from impaired signal transduction), which decreases subsequent calcium release by the SR. There can also be a decrease in TN-C affinity for calcium, so that an increase in free calcium near the troponin complex has less of an activating effect on cardiac contraction. In some forms of diastolic heart failure, there is evidence that the function of the SR ATP-dependent calcium pump is impaired. This defect would slow the rate of calcium uptake by the SR and reduce the rate of relaxation, leading to diastolic dysfunction.
Revised 11/04/2023