 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Circulating Catecholamines
Circulating catecholamines, epinephrine and norepinephrine, originate from two sources. Epinephrine is released by the adrenal medulla upon activation of preganglionic sympathetic nerves innervating this tissue. This activation occurs during times of stress (e.g., exercise, heart failure, hemorrhage, emotional stress or excitement, pain). Norepinephrine is also released by the adrenal medulla (approximately 20% of its total catecholamine release is norepinephrine); however, the primary source of circulating norepinephrine is spillover from sympathetic nerves innervating blood vessels. Normally, most of the norepinephrine released by sympathetic nerves is taken back up by the nerves (a small fraction is also taken up by extra-neuronal tissues) where it is metabolized. A small amount of norepinephrine that is recycled and metabolized diffuses into the blood and circulates throughout the body. At times of high sympathetic nerve activation, the amount of norepinephrine entering the blood increases dramatically.
There is also a specific adrenal medullary disorder (chromaffin cell tumor; pheochromocytoma) that causes very high circulating levels of catecholamines. This can lead to a hypertensive crisis.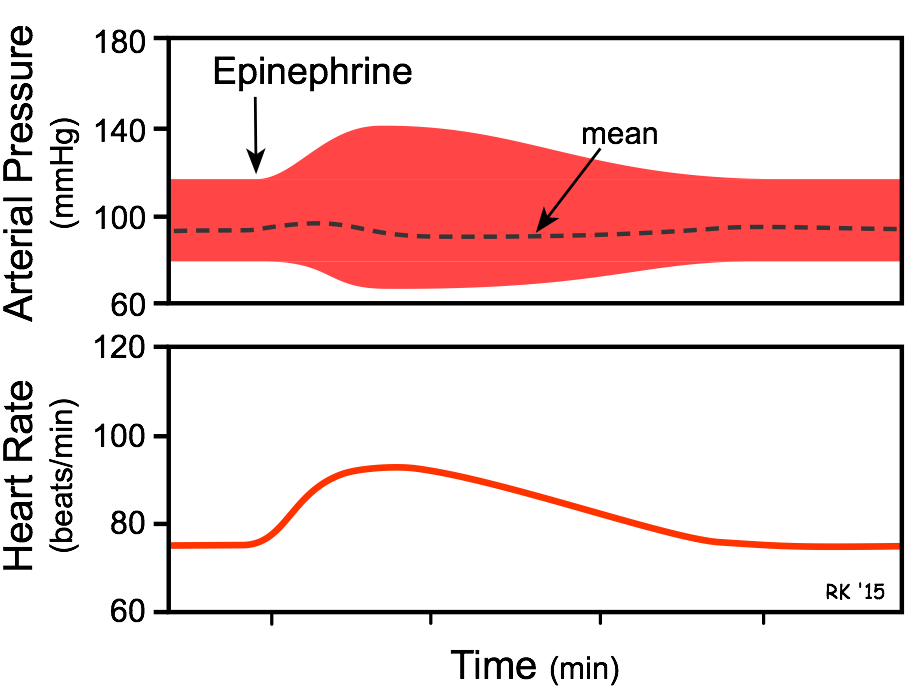
Circulating Epinephrine Causes:
- Increased heart rate and inotropy (β1-adrenoceptor mediated).
- Vasoconstriction of systemic arteries and veins via postjunctional α 1 and α 2 adrenoceptors.
- Vasodilation in muscle and liver vasculatures at low concentrations (β2-adrenoceptor); vasoconstriction at high concentrations (α-adrenoceptor mediated).
- The overall cardiovascular response to low-to-moderate circulating concentrations of epinephrine is increased cardiac output and a redistribution of the cardiac output to muscular and hepatic circulations, with only a small change in mean arterial pressure. Although cardiac output is increased, arterial pressure does not change much because the systemic vascular resistance falls due to the activation of vascular β2-adrenoceptors. At high plasma concentrations, epinephrine increases arterial pressure (not shown in figure) because of binding to α-adrenoceptors on blood vessels, which offsets the β2-adrenoceptor mediated vasodilation.
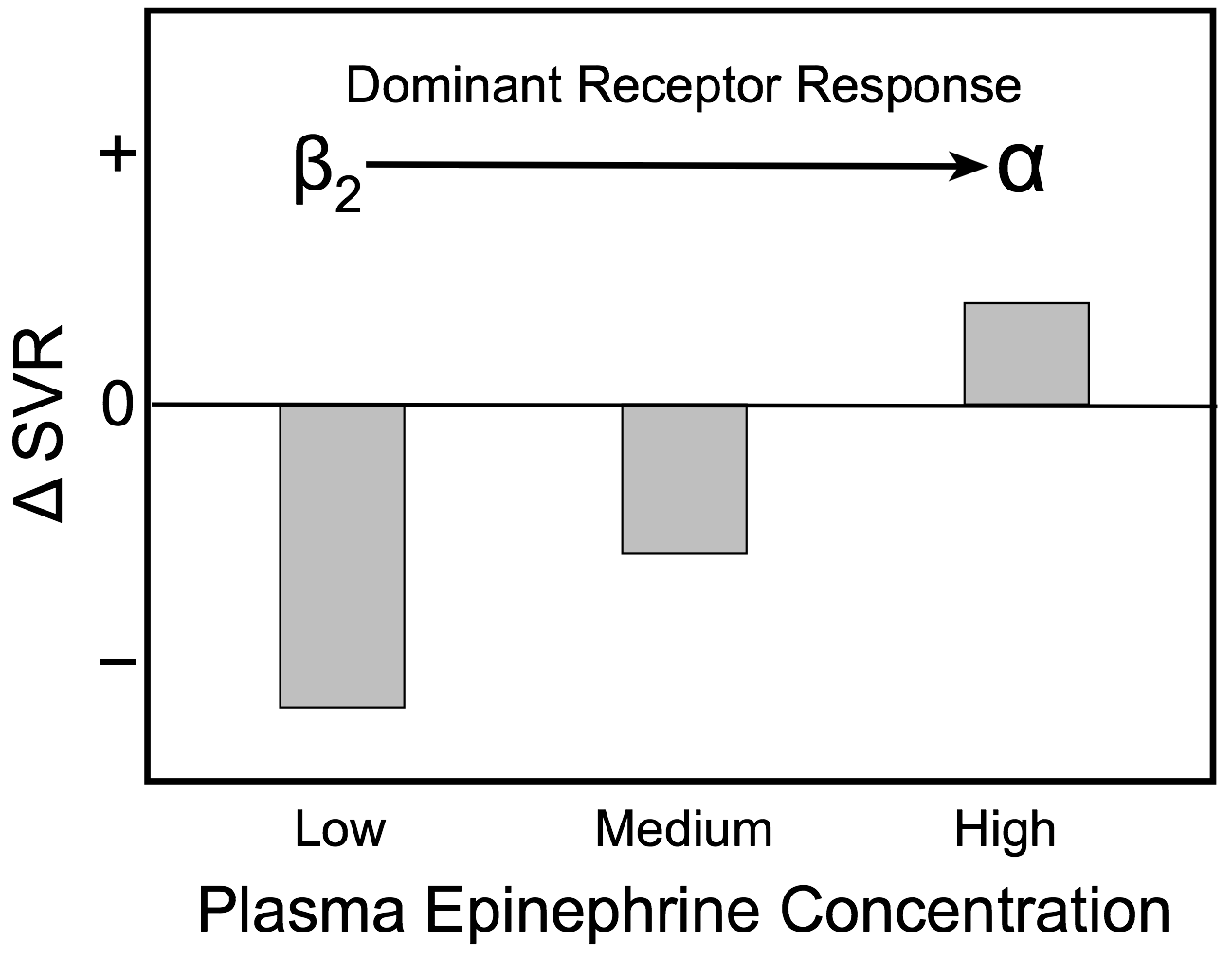 The effects of low, medium and high plasma concentrations of epinephrine on systemic vascular resistance are shown in the bar graph. At low plasma levels, epinephrine preferentially binds to high affinity vascular β2-adrenoceptors and causes vasodilation, which results in a fall in systemic vascular resistance. As the concentration of epinephrine increases, lower affinity α-adrenoceptors begin to bind epinephrine, which partially offsets the β2-adrenoceptor-mediated vasodilatory effects of epinephrine. At high circulating concentrations, more α-adrenoceptors are bound to epinephrine and the balance of vasodilatory and vasoconstrictor actions of epinephrine shifts to net vasoconstriction (increased systemic vascular resistance).
The effects of low, medium and high plasma concentrations of epinephrine on systemic vascular resistance are shown in the bar graph. At low plasma levels, epinephrine preferentially binds to high affinity vascular β2-adrenoceptors and causes vasodilation, which results in a fall in systemic vascular resistance. As the concentration of epinephrine increases, lower affinity α-adrenoceptors begin to bind epinephrine, which partially offsets the β2-adrenoceptor-mediated vasodilatory effects of epinephrine. At high circulating concentrations, more α-adrenoceptors are bound to epinephrine and the balance of vasodilatory and vasoconstrictor actions of epinephrine shifts to net vasoconstriction (increased systemic vascular resistance).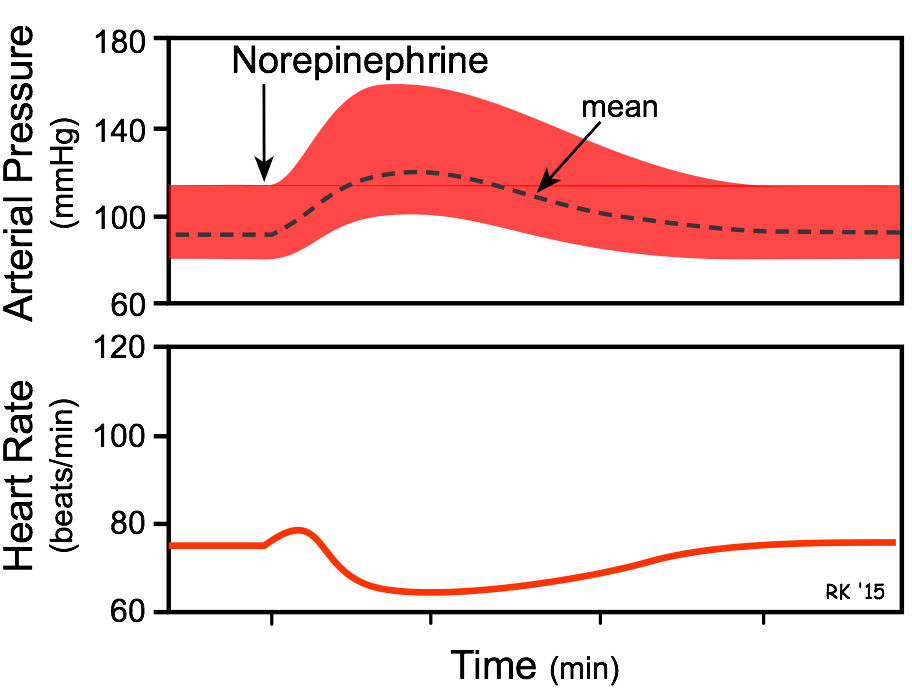
Circulating Norepinephrine Causes:
- Increased heart rate (although only transient) and increased inotropy (β1-adrenoceptor mediated) are the direct effects norepinephrine on the heart.
- Vasoconstriction occurs in most systemic arteries and veins (postjunctional α 1 and α 2 adrenoceptors).
- The overall cardiovascular response is increased cardiac output and systemic vascular resistance, which results in an elevation in arterial blood pressure. Heart rate, although initially directly stimulated by norepinephrine, decreases because of baroreflex activation, causing vagal-mediated slowing of the heart in response to the elevation in arterial pressure.
Pharmacologic Blocking of the Actions of Circulating Catecholamines
Because catecholamines act on the heart and blood vessels through alpha and beta adrenoceptors, the cardiovascular actions of catecholamines can be blocked by treatment with alpha-blockers and beta-blockers. Blocking either the alpha or beta adrenoceptor alone alters the response of the catecholamine because the other adrenoceptor will still bind to the catecholamine. For example, if a moderate dose of epinephrine is administered in the presence of alpha-adrenoceptor blockade, vascular β2-adrenoceptor activation unopposed by vascular α-adrenoceptor activation will cause a large hypotensive response because of systemic vasodilation despite the cardiac stimulation that occurs through β1-adrenoceptor activation.
Revised 12/7/2022