 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Regulation of Pacemaker Activity
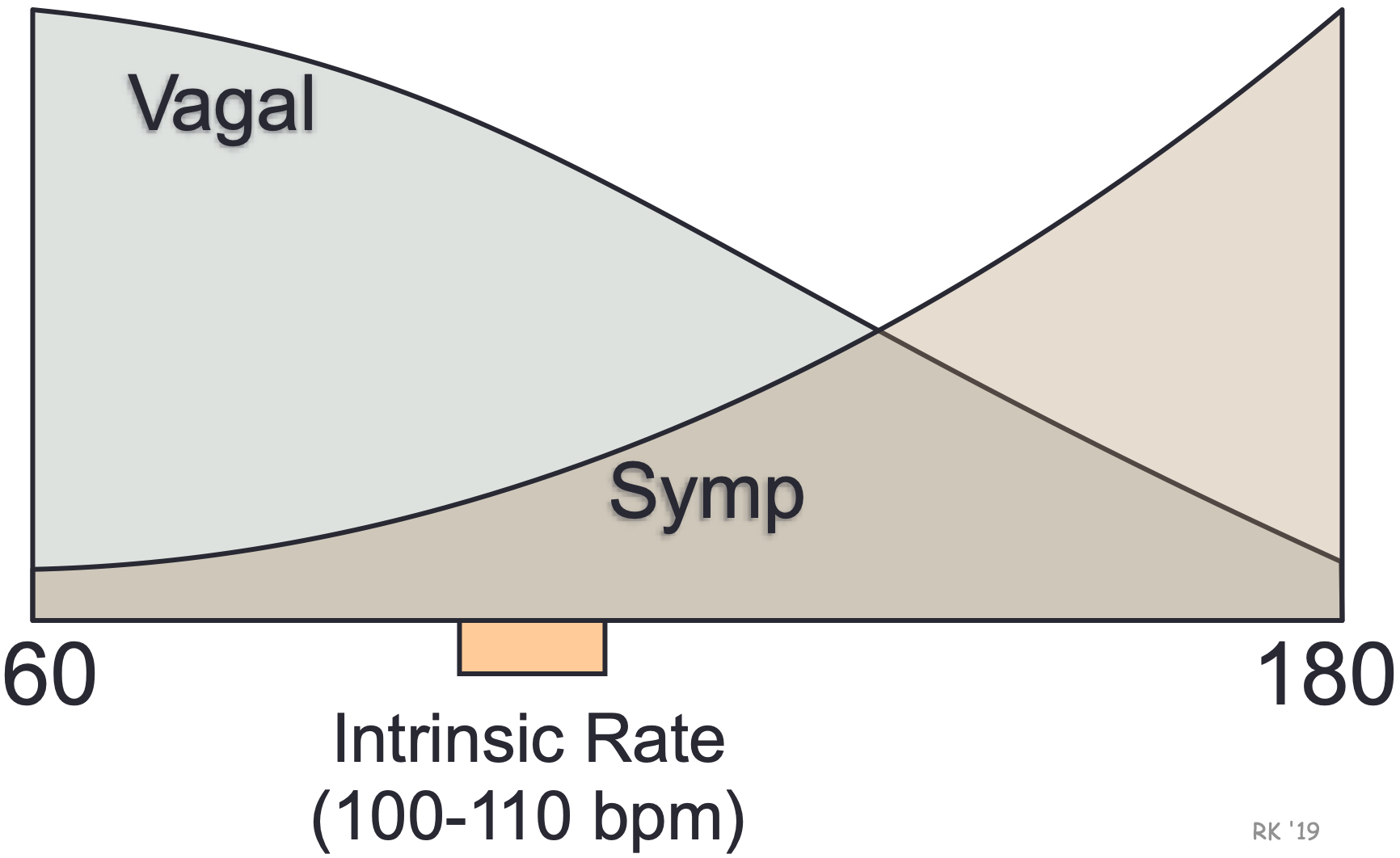 The SA node displays intrinsic automaticity (spontaneous pacemaker activity) at a rate of 100-110 action potentials (“beats”) per minute. This intrinsic rhythm is primarily influenced by autonomic nerves, with vagal influences being dominant over sympathetic influences at rest. This “vagal tone” reduces the resting heart rate down to 60-80 beats/min. The SA node is predominantly innervated by efferent branches of the right vagus nerves, although some innervation from the left vagus is often observed. Experimental denervation of the right vagus to the heart leads to an abrupt increase in SA nodal firing rate if the resting heart rate is below 100 beats/min. A similar response is noted when a drug, such as atropine, is administered. This drug blocks vagal influences at the SA node by antagonizing the muscarinic receptors that bind to acetylcholine, which is the neurotransmitter released by the vagus nerve. For the heart rate to increase during physical activity, the medullary centers controlling autonomic function reduce vagal efferent activity and increase sympathetic efferent activity to the SA node. High heart rates cannot be achieved in the absence of vagal inhibition.
The SA node displays intrinsic automaticity (spontaneous pacemaker activity) at a rate of 100-110 action potentials (“beats”) per minute. This intrinsic rhythm is primarily influenced by autonomic nerves, with vagal influences being dominant over sympathetic influences at rest. This “vagal tone” reduces the resting heart rate down to 60-80 beats/min. The SA node is predominantly innervated by efferent branches of the right vagus nerves, although some innervation from the left vagus is often observed. Experimental denervation of the right vagus to the heart leads to an abrupt increase in SA nodal firing rate if the resting heart rate is below 100 beats/min. A similar response is noted when a drug, such as atropine, is administered. This drug blocks vagal influences at the SA node by antagonizing the muscarinic receptors that bind to acetylcholine, which is the neurotransmitter released by the vagus nerve. For the heart rate to increase during physical activity, the medullary centers controlling autonomic function reduce vagal efferent activity and increase sympathetic efferent activity to the SA node. High heart rates cannot be achieved in the absence of vagal inhibition.
The rate of SA nodal firing can be altered by:
1. Changes in autonomic nerve activity (sympathetic and vagal)
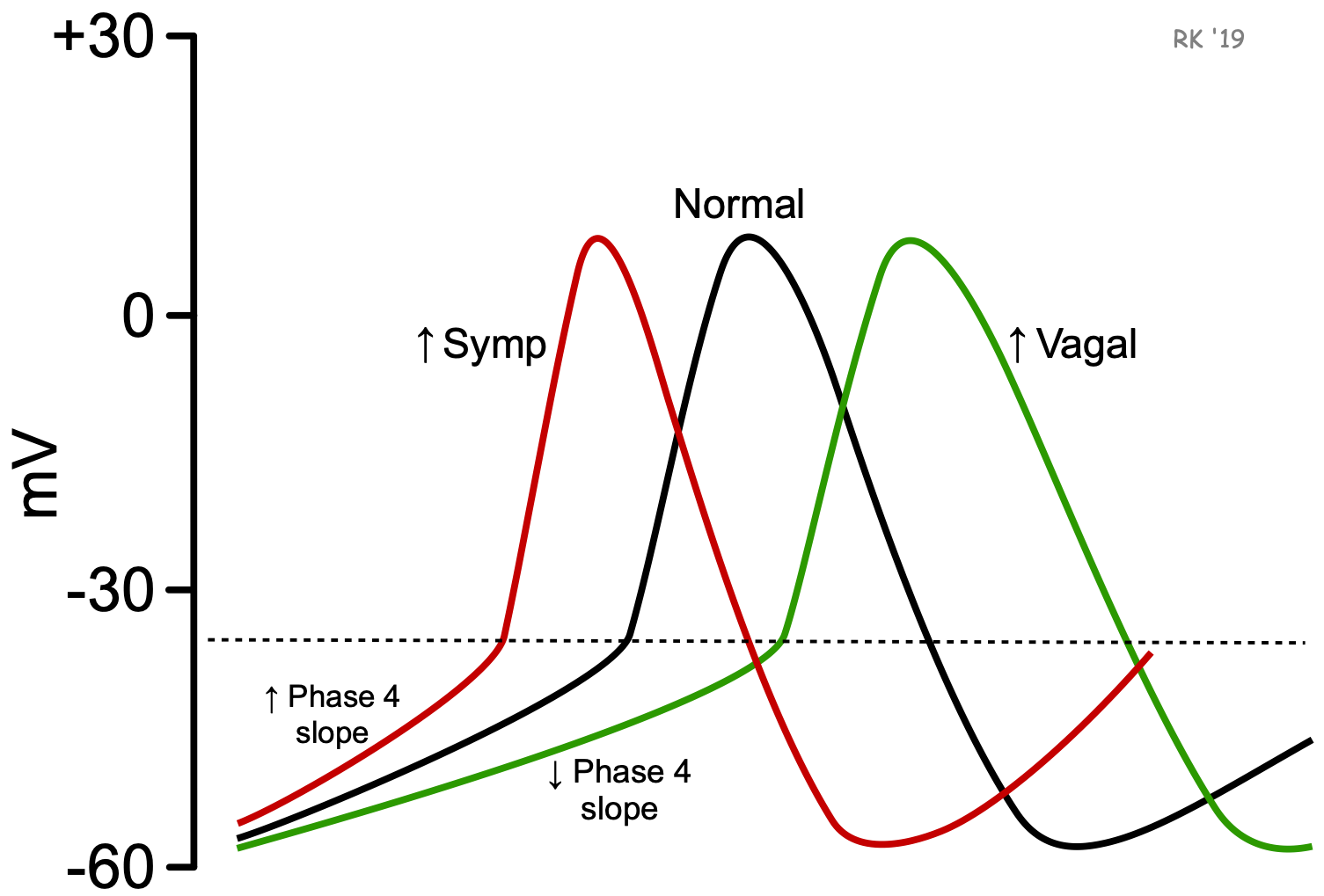
To increase heart rate, the autonomic nervous system increases sympathetic outflow to the SA node, with concurrent inhibition of vagal tone. Inhibition of vagal tone is necessary for the sympathetic nerves to increase heart rate because vagal influences inhibit the action of sympathetic nerve activity at the SA node.
Norepinephrine, released by sympathetic activation of the SA node, binds to beta-adrenoceptors. This increases the rate of pacemaker firing primarily by increasing the slope of phase 4, which decreases the time required to reach the threshold voltage. This occurs by increasing If (“funny” pacemaker currents) and increasing slow inward Ca++ currents. Parasympathetic (vagal) activation, which releases acetylcholine onto the SA node that binds to muscarinic receptors, decreases pacemaker rate (phase 4 slope) by increasing potassium conductance and decreasing the pacemaker currents (If) and slow inward calcium currents. These changes in ion currents decrease the slope of phase 4 of the action potential, thereby increasing the time required to reach the threshold voltage. Vagal activity also hyperpolarizes the pacemaker cell during Phase 4, which contributes to the longer time to reach threshold voltage.
2. Circulating hormones
Pacemaker activity is also altered by hormones. For example, hyperthyroidism induces tachycardia and hypothyroidism induces bradycardia. Circulating epinephrine causes tachycardia by a mechanism similar to norepinephrine released by sympathetic nerves.
3. Serum ion concentrations
Changes in the serum concentration of ions, particularly potassium, can cause changes in the SA nodal firing rate. Hyperkalemia induces bradycardia or can even stop SA nodal firing. Hyperkalemia causes membrane depolarization, which diminishes the degree of hyperpolarization that occurs at the end of phase 3. This prevents full reactivation of pacemaker channels for Na+ and Ca++ so that the slope of phase 4 is decreased. Hypokalemia increases the rate of phase 4 depolarization and causes tachycardia by enhancing phase 3 repolarization, which causes greater activation of channels responsible for the inward pacemaker currents. Note, however, that the effects of changes in serum potassium are complex, with multiple channels and ion pumps being affected by potassium concentration.
4. Cellular hypoxia
Cellular hypoxia (usually because of ischemia) depolarizes the membrane potential, causing bradycardia. Without adequate oxygen, ATP dependent ion pumps in the cell membrane cannot operate. This leads to cellular depolarization because of a loss of normal ion gradients (particularly K+) across the membrane. Because a hyperpolarized state at the end of phase 3 is necessary for pacemaker channels to become reactivated, pacemaker channels remain inactivated in depolarized cells. This suppresses pacemaker currents and decreases the slope of phase 4. This is one reason cellular hypoxia, which depolarizes the cell and alters phase 3 hyperpolarization, leads to a reduction in pacemaker rate (i.e., produces bradycardia). Severe hypoxia completely stops pacemaker activity.
5. Drugs
Various drugs used as antiarrhythmics also influence the SA nodal rhythm. Calcium-channel blockers, for example, cause bradycardia by inhibiting the slow inward Ca++ currents during phase 4 and phase 0. Drugs affecting autonomic control or autonomic receptors (e.g., beta-blockers, muscarinic antagonists) directly or indirectly alter pacemaker activity. Digoxin causes bradycardia by increasing parasympathetic (vagal) activity on the SA node; however, at toxic concentrations, digitalis increases automaticity and therefore can cause tachyarrhythmias. This toxic effect is related to the inhibitory effects of digoxin on the membrane Na+/K+-ATPase, which leads to cellular depolarization, increased intracellular calcium, and changes in ion conductances.
6. Age
Pacemaker activity is influenced dramatically by age. Many formulas have been developed to estimate the effects of age on maximal heart rate; however, the following simple formula is commonly used and serves to illustrate how age affects maximal heart rate:
HRmax = 220 — age in years
Although this formula is commonly used in the US health fitness industry, many studies have shown that this simple formula underestimates maximal HR by 10 or more beats/min in older individuals. A study done on over 3000 healthy men and women of a wide age range (Scand J Med Sci Sports 23:697-704, 2013) indicated that maximal HR is better estimated by 211 – (0.64 x age). The following comparison shows the differences in estimated maximal HR for 30- and 70-year-old individuals based on these two calculations:
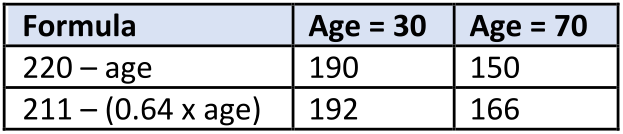
Therefore, maximal HR is reduced with increasing age, which also reduces maximal cardiac output during exertion. This effect is independent of gender, type of physical activity, maximal oxygen consumption, or body mass index. It is genetically determined and cannot be significantly modified by exercise training or by external factors.
Revised 12/03/2022