 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Cardiac Inotropy (Contractility)
Changes in inotropy are an important feature of cardiac muscle because, unlike skeletal muscle, cardiac muscle cannot modulate its force generation through changes in motor nerve activity and motor unit recruitment. When heart muscle contracts, all muscle fibers are activated and the only mechanisms that can alter force generation are changes in fiber length (preload; length-dependent activation) and changes in inotropy (length-independent activation). The effects of changes in inotropy on force generation can be illustrated by length-tension diagrams, in which increased inotropy increases active tension at a fixed preload. The inotropic property of cardiac muscle is displayed in the force-velocity relationship as a change in Vmax. A change in the maximal velocity of fiber shortening at zero afterload. The increased velocity of fiber shortening that occurs with increased inotropy increases the rate of ventricular pressure development, which is manifested as an increase in maximal dP/dt (i.e., rate of pressure change) measured during the phase of isovolumetric contraction. Because of these changes in the mechanical properties of contracting cardiac muscle, an increase in inotropy leads to an increase in ventricular stroke volume.
Effects of Inotropy on Frank-Starling Curves
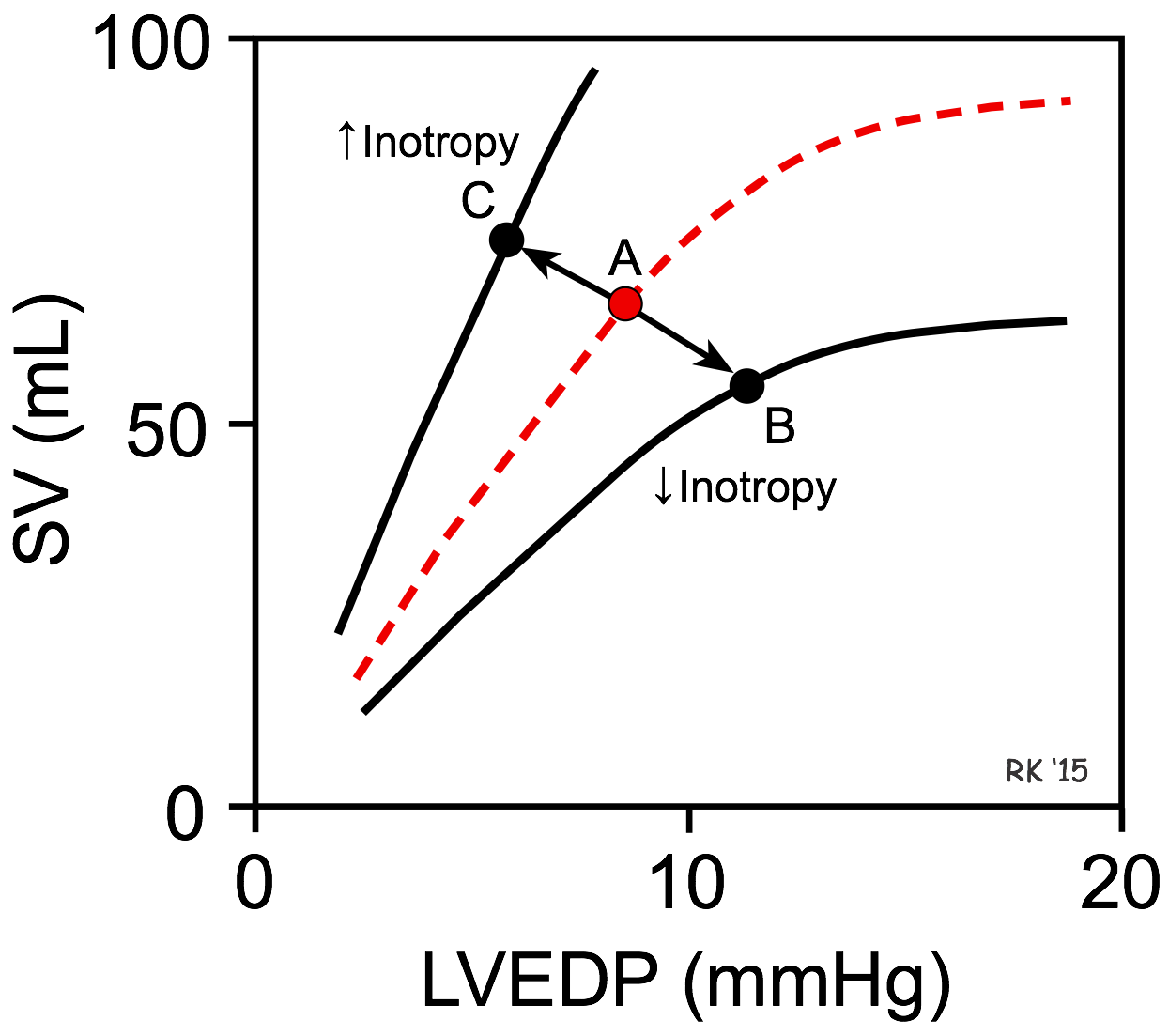 By altering the rate of ventricular pressure development, the rate of ventricular ejection into the aorta (i.e., ejection velocity) is changed. Because there is finite time available for ejection (~200 msec), changes in ejection velocity alter the stroke volume – increased ejection velocity increases stroke volume, whereas reduced ejection velocity decreases stroke volume.
By altering the rate of ventricular pressure development, the rate of ventricular ejection into the aorta (i.e., ejection velocity) is changed. Because there is finite time available for ejection (~200 msec), changes in ejection velocity alter the stroke volume – increased ejection velocity increases stroke volume, whereas reduced ejection velocity decreases stroke volume.
A decrease in inotropy shifts the Frank-Starling curve downward (point A to B in the figure). This causes the stroke volume (SV) to decrease and the left ventricular end-diastolic pressure (LVEDP) and volume to increase. The change in SV is the primary response, whereas the change in LVEDP is a secondary response to the change in SV. This occurs, for example, when there is a loss in ventricular inotropy during certain types of heart failure. If inotropy is increased (as occurs during exercise), the Frank-Starling curve shifts up and to the left (point A to C in the figure), resulting in an increase in SV and a decrease in LVEDP. Once a Frank-Starling curve shifts in response to an altered inotropic state, changes in ventricular filling will alter the SV by moving either up or down the new Frank-Starling curve.
Effects of Inotropy on Ventricular Pressure-Volume Loops
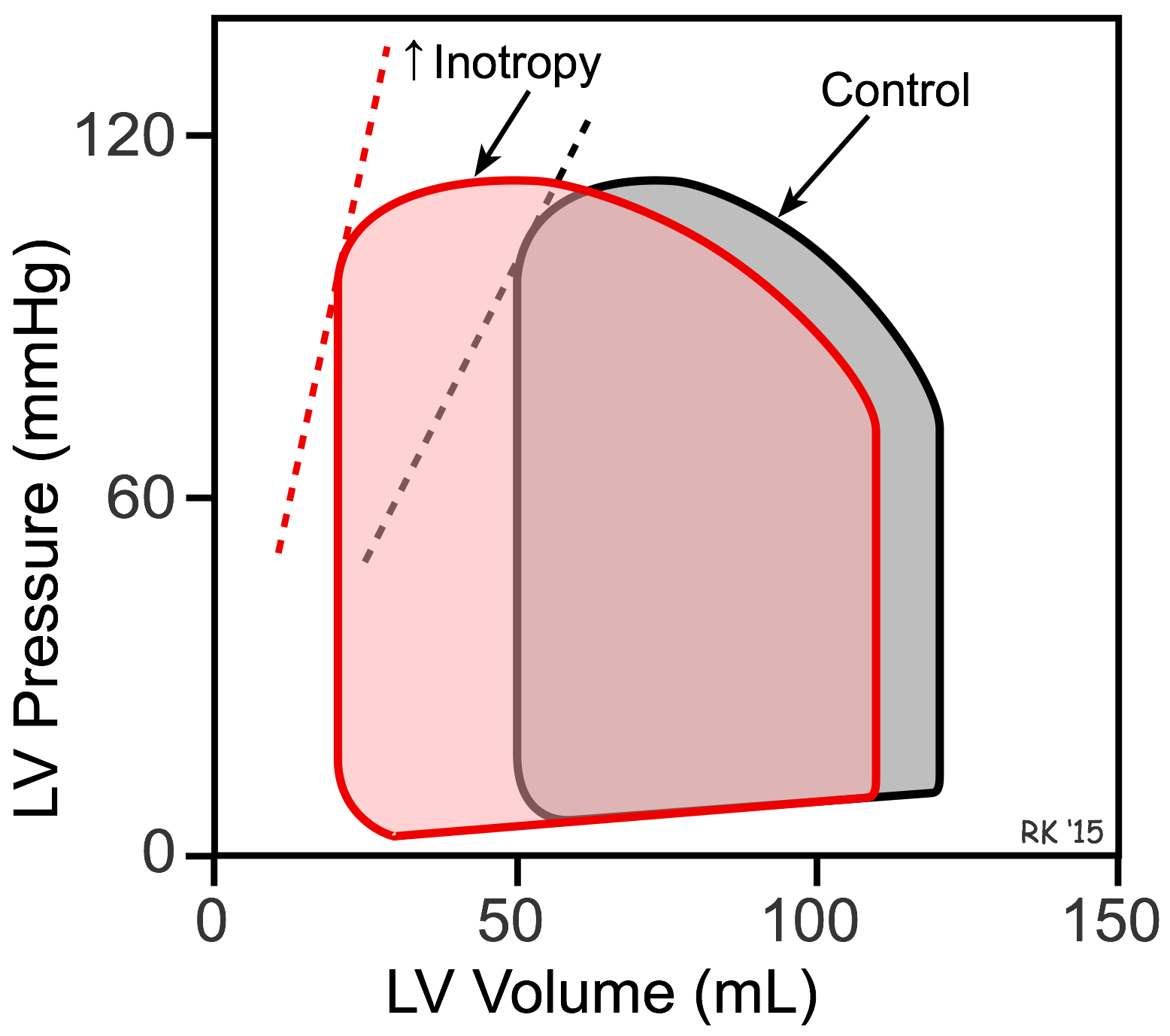 The reason LVEDP falls when SV is increased can best be shown using left ventricular (LV) pressure-volume loops (see figure). In this figure, the control loop has an end-diastolic volume of 120 mL and an end-systolic volume of 50 mL. The width of the loop (end-diastolic minus end-systolic volume) is the stroke volume (70 mL). When inotropy is increased (at constant arterial pressure and heart rate) SV increases, which reduces the end-systolic volume to 20 mL. This is accompanied by a secondary reduction in ventricular end-diastolic volume (to 110 mL) and pressure because when the SV is increased the ventricle contains less residual blood volume after ejection (decreased end-systolic volume) that can be added to the incoming venous return during filling. Therefore, ventricular filling (end-diastolic volume) is reduced. The dashed lines for the two loops represent the end-systolic pressure-volume relationship (ESPVR). The ESPVR is shifted to the left and its slope becomes steeper when inotropy is increased. The ESPVR is sometimes used as an index of ventricular inotropic state.
The reason LVEDP falls when SV is increased can best be shown using left ventricular (LV) pressure-volume loops (see figure). In this figure, the control loop has an end-diastolic volume of 120 mL and an end-systolic volume of 50 mL. The width of the loop (end-diastolic minus end-systolic volume) is the stroke volume (70 mL). When inotropy is increased (at constant arterial pressure and heart rate) SV increases, which reduces the end-systolic volume to 20 mL. This is accompanied by a secondary reduction in ventricular end-diastolic volume (to 110 mL) and pressure because when the SV is increased the ventricle contains less residual blood volume after ejection (decreased end-systolic volume) that can be added to the incoming venous return during filling. Therefore, ventricular filling (end-diastolic volume) is reduced. The dashed lines for the two loops represent the end-systolic pressure-volume relationship (ESPVR). The ESPVR is shifted to the left and its slope becomes steeper when inotropy is increased. The ESPVR is sometimes used as an index of ventricular inotropic state.
Changes in inotropy produce significant changes in ejection fraction (EF, calculated as stroke volume divided by end-diastolic volume). In the previous figure, the control EF is 0.58 and increases to 0.82 with increased inotropy. Therefore, increasing inotropy leads to an increase in EF. In contrast, decreasing inotropy decreases EF. Therefore, EF is frequently used as a clinical index to assess the inotropic state of the heart. In heart failure caused by systolic dysfunction, there is a decrease in inotropy that leads to a fall in stroke volume and a secondary increase in preload, decreasing EF.
Changes in inotropic state are important during exercise. Increases in inotropic state help to maintain stroke volume at high heart rates and elevated arterial pressures. Increased heart rate alone decreases stroke volume because of reduced time for diastolic filling, which decreases end-diastolic volume. Elevated arterial pressure during exercise increases afterload on the heart, which reduces stroke volume. When the inotropic state increases, end-systolic volume decreases so that stroke volume can be maintained and allowed to increase despite reduced time for ventricular filling and elevated arterial pressure.
Factors Regulating Inotropy
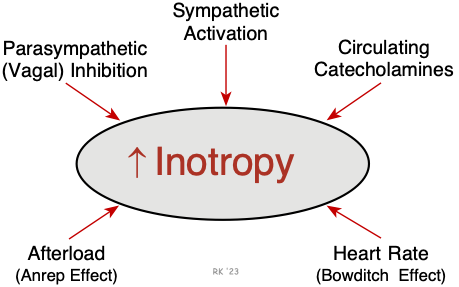 The most important mechanism regulating inotropy is the autonomic nerves. Sympathetic nerves play a prominent role in ventricular and atrial inotropic regulation, while parasympathetic nerves (vagal efferent nerves) have a significant negative inotropic effect in the atria but only a small effect in the ventricles. Under certain conditions (e.g., exercise, stress, and anxiety), high levels of circulating epinephrine augment sympathetic adrenergic effects. In the human heart, an abrupt increase in afterload can cause an increase in inotropy (Anrep effect). An increase in heart rate also stimulates inotropy (Bowditch effect; treppe; frequency-dependent inotropy). This latter phenomenon is probably because of an inability of the Na+/K+-ATPase to keep up with the sodium influx at higher heart rates, which leads to an accumulation of intracellular calcium via the sodium-calcium exchanger. Systolic failure that results from cardiomyopathy, ischemia, valve disease, arrhythmias, and other conditions is characterized by a loss of intrinsic inotropy.
The most important mechanism regulating inotropy is the autonomic nerves. Sympathetic nerves play a prominent role in ventricular and atrial inotropic regulation, while parasympathetic nerves (vagal efferent nerves) have a significant negative inotropic effect in the atria but only a small effect in the ventricles. Under certain conditions (e.g., exercise, stress, and anxiety), high levels of circulating epinephrine augment sympathetic adrenergic effects. In the human heart, an abrupt increase in afterload can cause an increase in inotropy (Anrep effect). An increase in heart rate also stimulates inotropy (Bowditch effect; treppe; frequency-dependent inotropy). This latter phenomenon is probably because of an inability of the Na+/K+-ATPase to keep up with the sodium influx at higher heart rates, which leads to an accumulation of intracellular calcium via the sodium-calcium exchanger. Systolic failure that results from cardiomyopathy, ischemia, valve disease, arrhythmias, and other conditions is characterized by a loss of intrinsic inotropy.
Besides these physiological mechanisms, various inotropic drugs are used clinically to stimulate the heart, particularly in acute and occasionally in chronic heart failure. These drugs include digoxin (inhibits sarcolemmal Na+/K+-ATPase), beta-adrenoceptor agonists (e.g., dopamine, dobutamine, epinephrine, isoproterenol), and phosphodiesterase inhibitors (e.g., milrinone).
Mechanisms of Inotropy
Most of the signal transduction pathways that stimulate inotropy ultimately involve Ca++, either by increasing Ca++ influx (via Ca++ channels) during the action potential (primarily during phase 2), by increasing the release of Ca++ by the sacroplasmic reticulum, or by sensitizing troponin-C (TN-C) to Ca++.
Revised 11/04/2023