 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Cardiac Afterload
What is Afterload?
Afterload can be thought of as the "load" that the heart must eject blood against. In simple terms, the afterload of the left ventricle is closely related to the aortic pressure. The afterload on individual muscle fibers within the wall of the heart is often expressed as ventricular wall stress (σ) and described by the following equation:
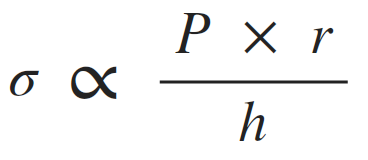
(P, ventricular pressure; r, ventricular radius; h, wall thickness).
This relationship is similar to the Law of LaPlace, which states that wall tension (T) is proportionate to the pressure (P) times radius (r) for thin-walled spheres or cylinders. Therefore, wall stress is wall tension divided by wall thickness. The exact equation depends on the cardiac chamber shape, which changes during the cardiac cycle; therefore, a single geometric relationship is sometimes assumed for simplicity. The above relationship is expressed as a proportionality to highlight how pressure, radius and wall thickness contribute to afterload.
The pressure that the left ventricle generates during systolic ejection is very close to aortic pressure unless aortic stenosis is present, in which case the left ventricular pressure during ejection can be much greater than aortic pressure. At a given intraventricular pressure, wall stress and therefore afterload are increased by an increase in ventricular chamber radius (ventricular dilation). A hypertrophied ventricle, which has a thickened wall, has less wall stress and reduced afterload. Hypertrophy, therefore, can be thought of as a mechanism that permits more parallel muscle fibers (actually, sarcomere units) to share in the wall tension that is determined at a given pressure and radius. The thicker the wall, the less tension experienced by each sarcomere unit.
Afterload on the left ventricle is increased when aortic pressure and systemic vascular resistance are increased, by aortic valve stenosis, and by ventricular dilation. When afterload increases, there is an increase in end-systolic volume and a decrease in stroke volume, as described below.
How Afterload Affects Stroke Volume and Preload
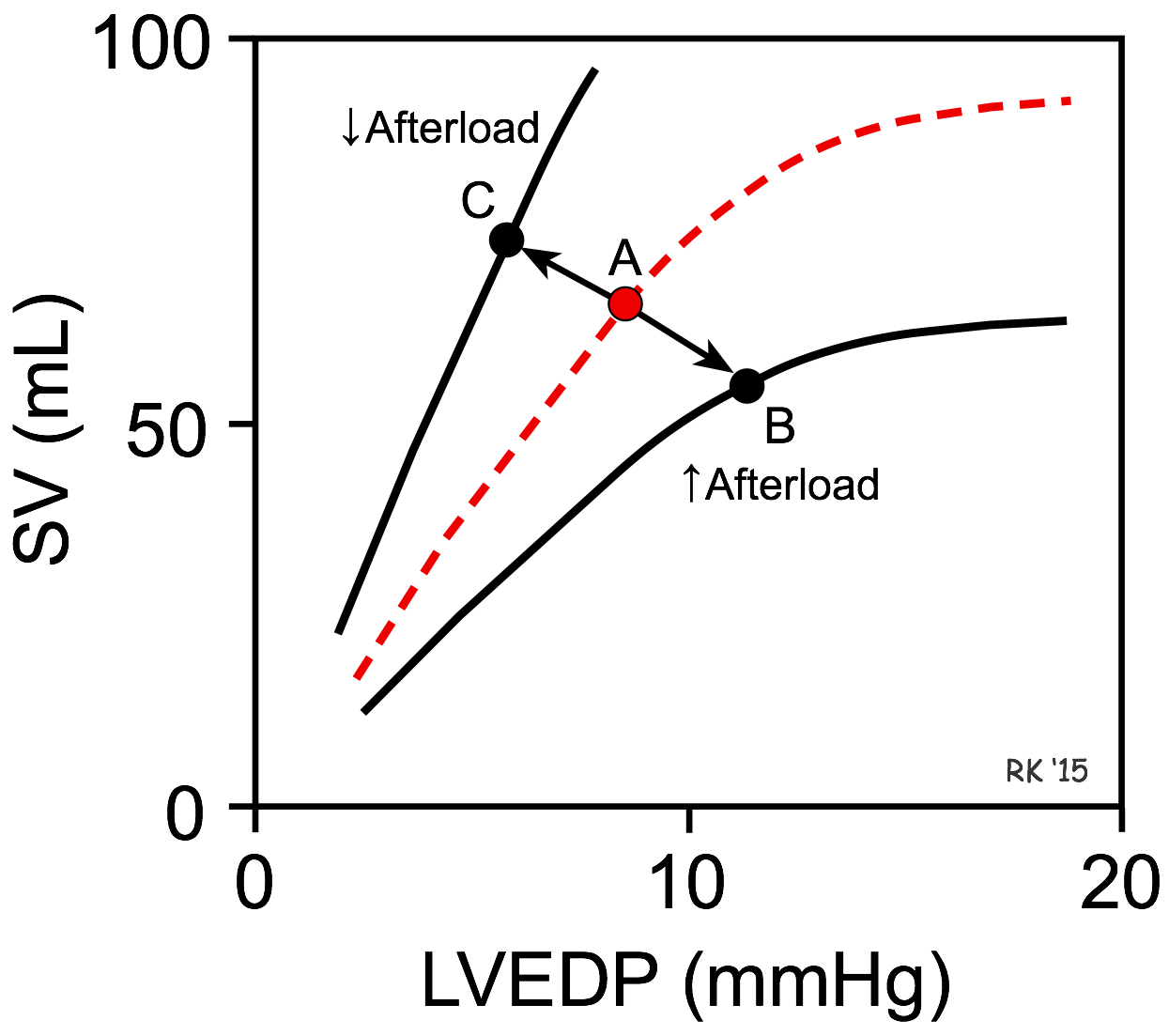 As shown in the figure, an increase in afterload shifts the Frank-Starling curve down and to the right (from point A to B), which decreases stroke volume (SV) and increases left ventricular end-diastolic pressure (LVEDP). The basis for this is found in the force-velocity relationship for cardiac myocytes. Briefly, an increase in afterload decreases the velocity of fiber shortening. Because the time available for ejection is finite (~200 msec), a decrease in fiber shortening velocity reduces the rate of volume ejection so that more blood is left within the ventricle at the end of systole (increased end-systolic volume). In contrast, a decrease in afterload shifts the Frank-Starling curve up and to the left (A to C), which increases SV and reduces LVEDP.
As shown in the figure, an increase in afterload shifts the Frank-Starling curve down and to the right (from point A to B), which decreases stroke volume (SV) and increases left ventricular end-diastolic pressure (LVEDP). The basis for this is found in the force-velocity relationship for cardiac myocytes. Briefly, an increase in afterload decreases the velocity of fiber shortening. Because the time available for ejection is finite (~200 msec), a decrease in fiber shortening velocity reduces the rate of volume ejection so that more blood is left within the ventricle at the end of systole (increased end-systolic volume). In contrast, a decrease in afterload shifts the Frank-Starling curve up and to the left (A to C), which increases SV and reduces LVEDP.
Afterload per se does not alter preload; however, preload changes secondarily to changes in afterload. Increasing afterload not only reduces stroke volume, but it also increases left ventricular end-diastolic pressure (LVEDP) (i.e., increases preload). This occurs because the increase in end-systolic volume (residual volume remaining in the ventricle after ejection) is added to the venous return into the ventricle, and this increases end-diastolic volume. This increase in preload activates the Frank-Starling mechanism, which partially compensates for the reduction in stroke volume caused by the increase in afterload.
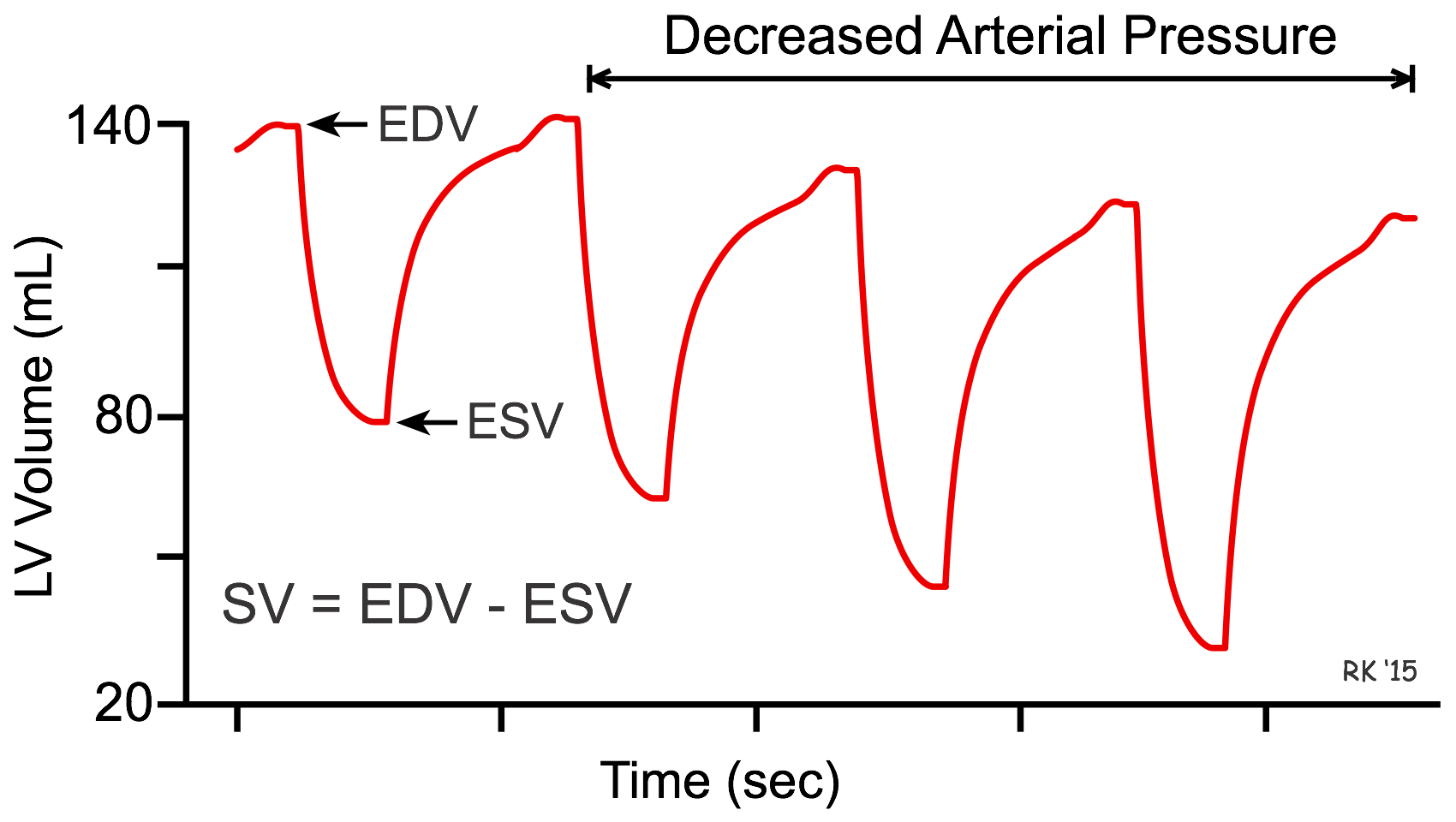 The interaction between afterload and preload is used in the treatment of heart failure, in which vasodilator drugs are used to augment stroke volume by decreasing arterial pressure (afterload), and to reduce ventricular preload. This can be illustrated by seeing how ventricular volume changes in response to a decrease in arterial pressure over several heart beats (see figure). In this example, the initial end-diastolic volume (EDV) is 140 mL and end-systolic volume (ESV) is 80 mL. When arterial pressure is reduced, the ventricle can eject blood more rapidly, which increases the stroke volume (difference between EDV and ESV) and decreases the ESV. Because less blood remains in the ventricle after systole, the ventricle does not fill to the same EDV found before the afterload reduction. Therefore, EDV (preload) is reduced as ESV decreases. Stroke volume increases overall because the reduction in EDV is less than the reduction in ESV.
The interaction between afterload and preload is used in the treatment of heart failure, in which vasodilator drugs are used to augment stroke volume by decreasing arterial pressure (afterload), and to reduce ventricular preload. This can be illustrated by seeing how ventricular volume changes in response to a decrease in arterial pressure over several heart beats (see figure). In this example, the initial end-diastolic volume (EDV) is 140 mL and end-systolic volume (ESV) is 80 mL. When arterial pressure is reduced, the ventricle can eject blood more rapidly, which increases the stroke volume (difference between EDV and ESV) and decreases the ESV. Because less blood remains in the ventricle after systole, the ventricle does not fill to the same EDV found before the afterload reduction. Therefore, EDV (preload) is reduced as ESV decreases. Stroke volume increases overall because the reduction in EDV is less than the reduction in ESV.
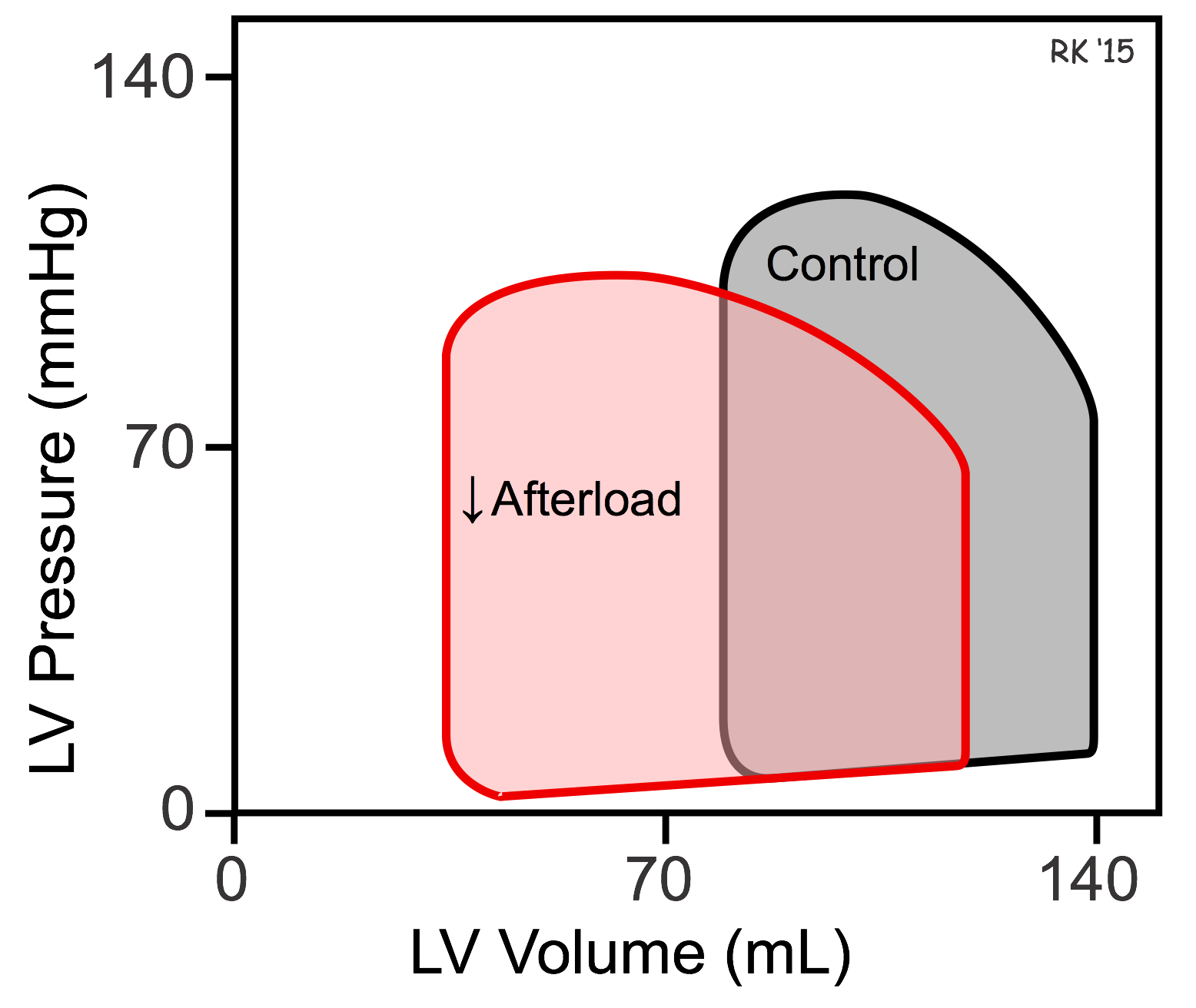 The effects of afterload on ventricular ESV and EDV can be illustrated using pressure-volume loops (see figure). If afterload is decreased by decreasing arterial pressure, as in the example discussed above, the ventricle needs to generate less pressure before the aortic valve opens. The ejection velocity after the valve opens is increased because decreased afterload increases the velocity of cardiac fiber shortening, as described by the force-velocity relationship. More blood is ejected (increased stroke volume), which decreases the ventricular ESV as shown in the pressure-volume loop. Because end-systolic volume is decreased, there is less blood within the ventricle to be added to the venous return, which decreases EDV. Ordinarily, in the final steady-state (after several beats), the decrease in EDV is less than the decrease in ESV so that the difference between the two, the stroke volume, is increased (i.e., the width of the pressure-volume loop is increased).
The effects of afterload on ventricular ESV and EDV can be illustrated using pressure-volume loops (see figure). If afterload is decreased by decreasing arterial pressure, as in the example discussed above, the ventricle needs to generate less pressure before the aortic valve opens. The ejection velocity after the valve opens is increased because decreased afterload increases the velocity of cardiac fiber shortening, as described by the force-velocity relationship. More blood is ejected (increased stroke volume), which decreases the ventricular ESV as shown in the pressure-volume loop. Because end-systolic volume is decreased, there is less blood within the ventricle to be added to the venous return, which decreases EDV. Ordinarily, in the final steady-state (after several beats), the decrease in EDV is less than the decrease in ESV so that the difference between the two, the stroke volume, is increased (i.e., the width of the pressure-volume loop is increased).
It is important to note that changes brought about by altered afterload are modified acutely by baroreceptor reflexes that alter heart rate and inotropy, both of which will modify the changes in EDV, ESV and stroke volume initiated by the change in afterload. For example, suddenly reducing afterload by decreasing arterial pressure will lead to a reflex increase in heart rate and inotropy. Increased heart rate, by reducing filling time, will further decrease in EDV and attenuate the stroke volume increase produced by reducing the afterload. Increased inotropy would tend to further reduce the ESV, further increase the SV, and further reduce EDV. The final steady-state response will be determined by the sum of the individual, yet interdependent, responses.
Revised 01/22/2023