 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Frank-Starling Mechanism
As described elsewhere, cardiac output increases or decreases in response to changes in heart rate or stroke volume. When a person stands up, for example, cardiac output falls because a fall in central venous pressure leads to a decrease in stroke volume. As another example, limb movement (muscle pump) during exercise enhances venous return to the heart, which causes an increase in stroke volume. What are the mechanisms by which changes in venous return alter stroke volume?
Venous Return and Stroke Volume
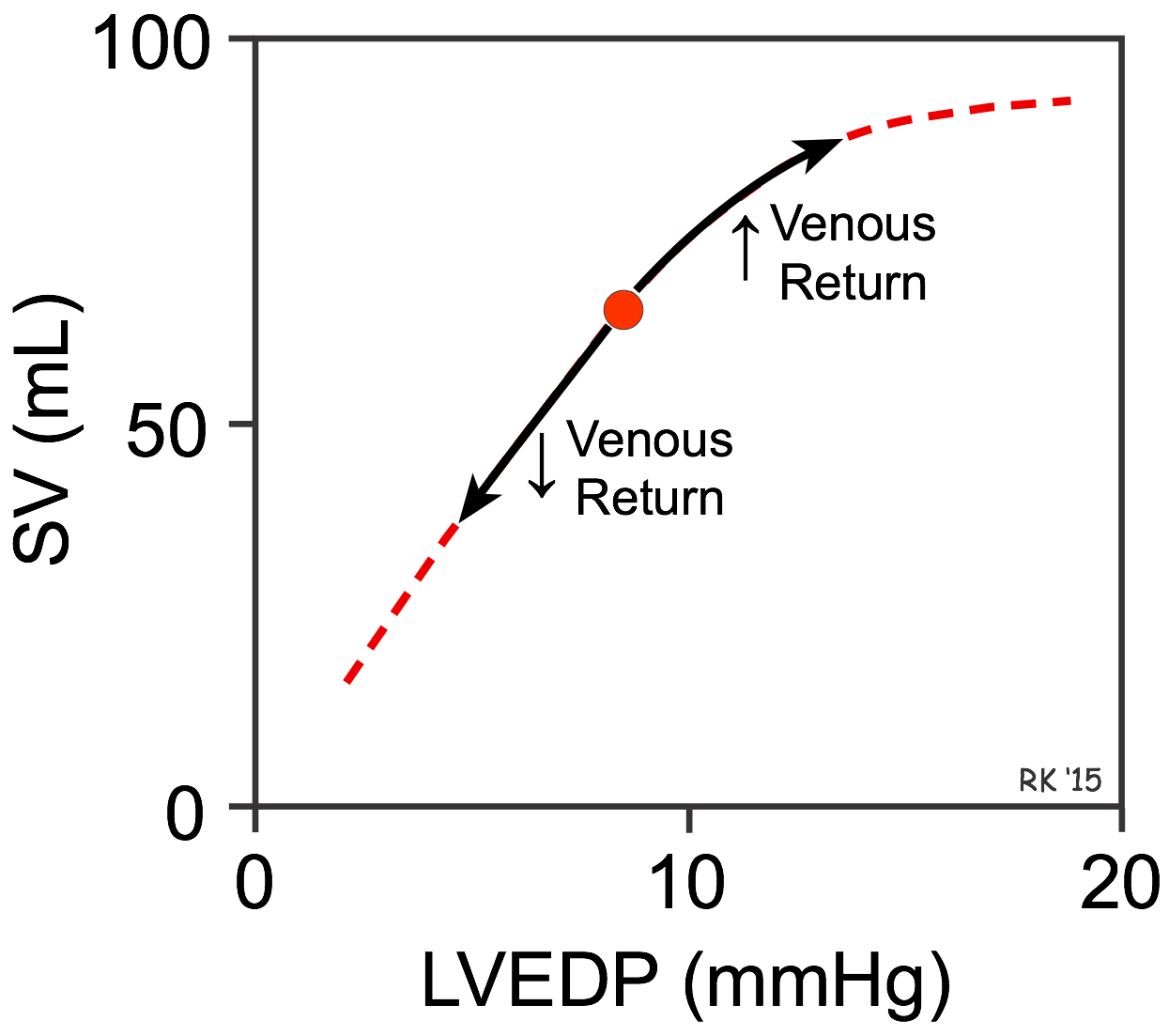 In the late19th century, Otto Frank found using isolated frog hearts that the strength of ventricular contraction was increased when the ventricle was stretched before contraction. This observation was extended by the elegant studies of Ernest Starling and colleagues in the early 20th century, who found that increasing venous return to the heart (see figure), which increased the filling pressure (left ventricular end-diastolic pressure; LVEDP in the figure) of the ventricle caused the stroke volume (SV) to increase. Conversely, decreasing venous return decreased stroke volume. This cardiac response to changes in venous return and ventricular filling pressure is intrinsic to the heart and does not depend on extrinsic neurohumoral mechanisms, although such mechanisms can modify the intrinsic cardiac response. In honor of these two early pioneers, the ability of the heart to change its force of contraction and therefore stroke volume in response to changes in venous return is called the Frank-Starling mechanism (or Starling's Law of the heart).
In the late19th century, Otto Frank found using isolated frog hearts that the strength of ventricular contraction was increased when the ventricle was stretched before contraction. This observation was extended by the elegant studies of Ernest Starling and colleagues in the early 20th century, who found that increasing venous return to the heart (see figure), which increased the filling pressure (left ventricular end-diastolic pressure; LVEDP in the figure) of the ventricle caused the stroke volume (SV) to increase. Conversely, decreasing venous return decreased stroke volume. This cardiac response to changes in venous return and ventricular filling pressure is intrinsic to the heart and does not depend on extrinsic neurohumoral mechanisms, although such mechanisms can modify the intrinsic cardiac response. In honor of these two early pioneers, the ability of the heart to change its force of contraction and therefore stroke volume in response to changes in venous return is called the Frank-Starling mechanism (or Starling's Law of the heart).
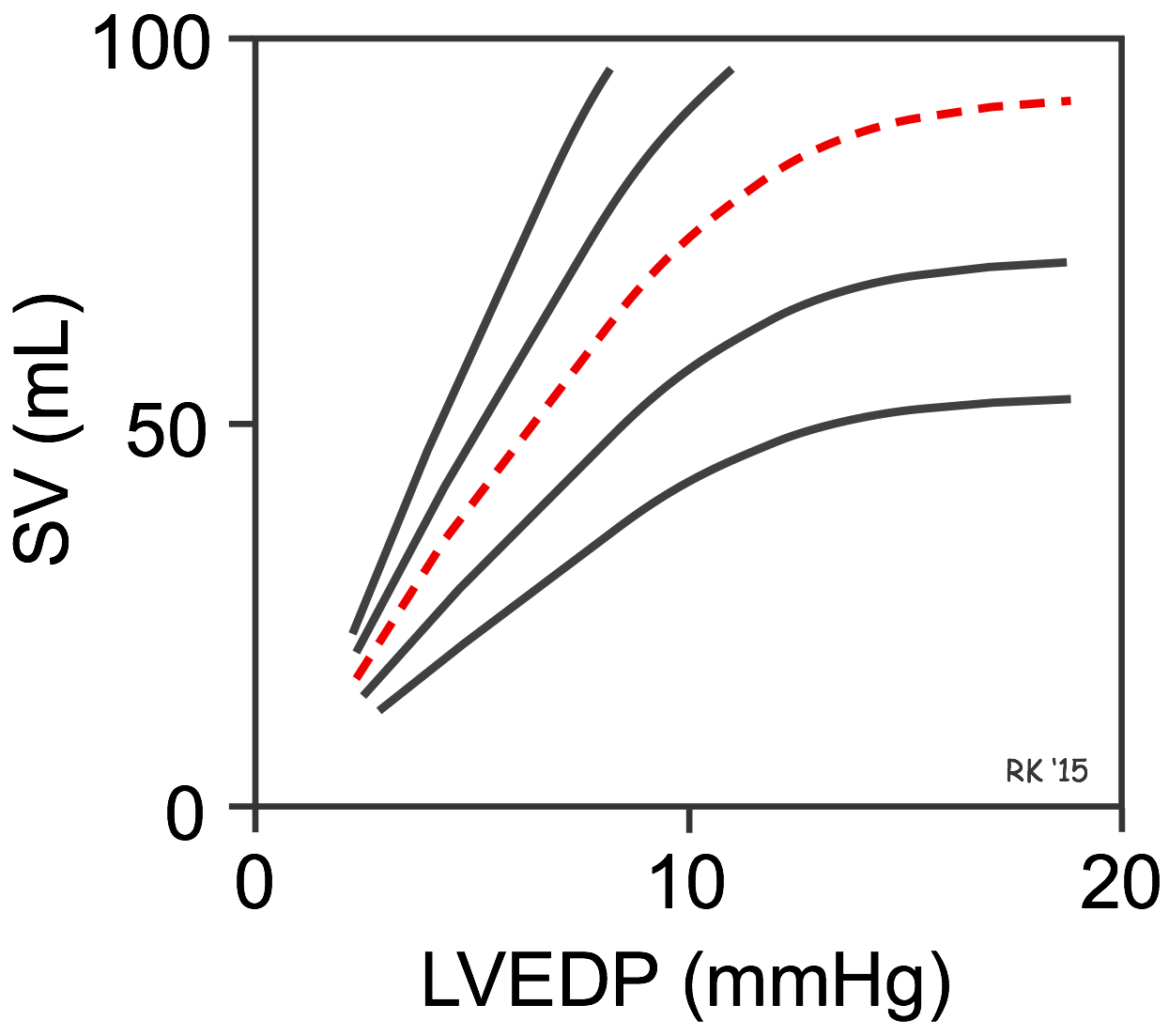 There is no single Frank-Starling curve on which the ventricle operates. Instead, there is a family of curves, each of which is defined by the afterload and inotropic state of the heart.
There is no single Frank-Starling curve on which the ventricle operates. Instead, there is a family of curves, each of which is defined by the afterload and inotropic state of the heart.
In the figure with multiple curves, the red dashed curve represents a "normal" ventricular Frank-Starling curve. Increasing afterload or decreasing inotropy shifts the curve down and to the right. Therefore, at a fixed LVEDP, depressing the curve will decrease the SV. Decreasing afterload and increasing inotropy shifts the curve up and to the left. Therefore, at a fixed LVEDP, shifting the Frank-Starling curve up and to the left increases SV at a fixed LVEDP. If ventricular inotropy and afterload do not change, the ventricle responds to changes in venous return and ventricular filling based on the unique curve for those conditions. In summary, changes in venous return cause the ventricle to move up or down along a single Frank-Starling curve; however, the slope of that curve is defined by the existing conditions of afterload and inotropy.
Frank-Starling curves show how changes in ventricular preload lead to changes in stroke volume. This type of graphical representation, however, does not show how changes in venous return affect end-diastolic and end-systolic volumes. To do this, it is necessary to describe ventricular function in terms of pressure-volume diagrams.
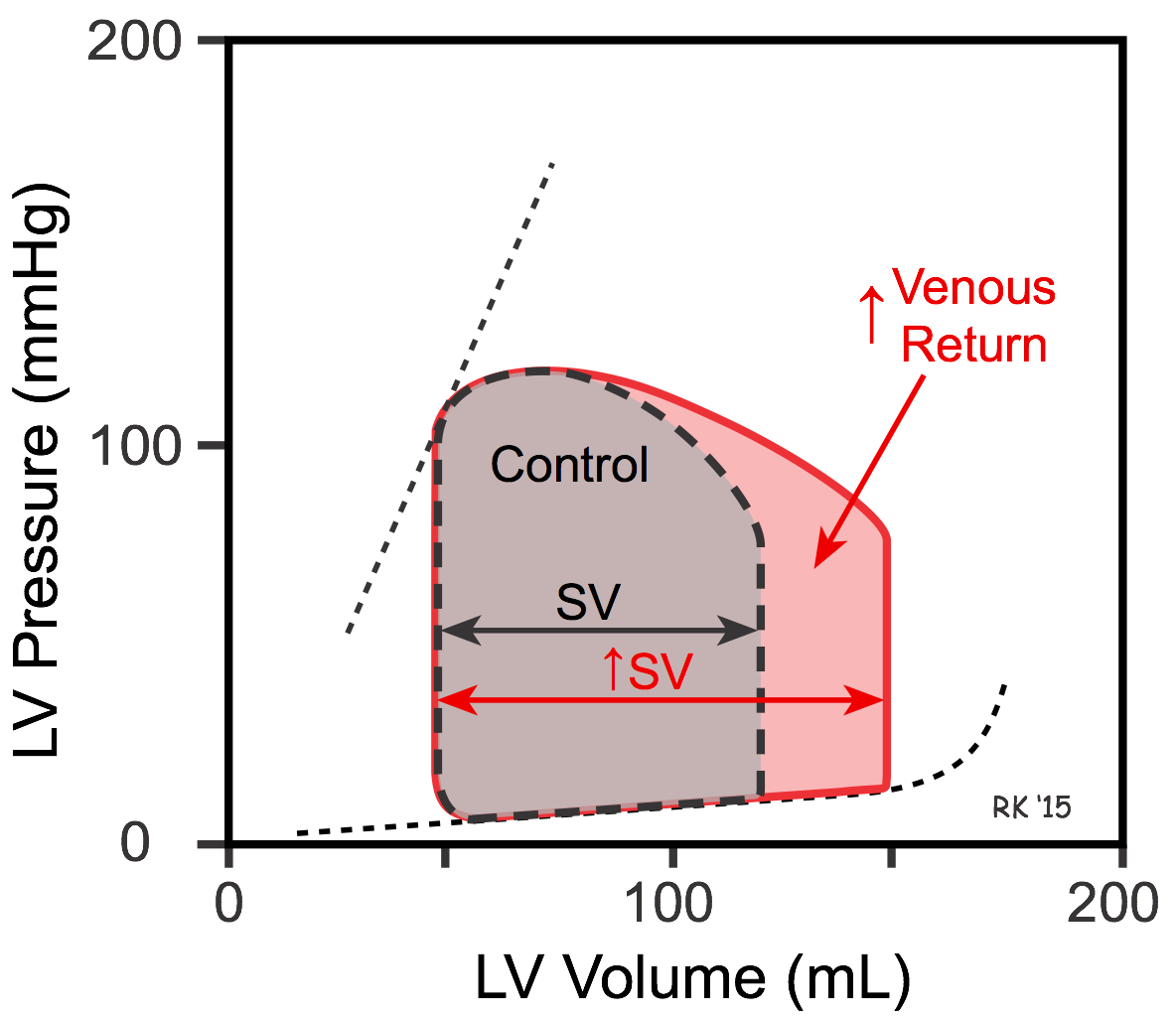 When venous return is increased, there is increased filling of the ventricle along its passive pressure filling curve, leading to an increase in end-diastolic volume (see Figure). If the ventricle now contracts at this increased preload, and the afterload and inotropy are held constant, the ventricle empties to the same end-systolic volume, increasing its stroke volume, which is defined as end-diastolic minus end-systolic volume. The increased stroke volume is represented as an increase in the width of the pressure-volume loop. The normal ventricle, therefore, can increase its stroke volume to match physiological increases in venous return. This is not, however, the case for ventricles that are in failure.
When venous return is increased, there is increased filling of the ventricle along its passive pressure filling curve, leading to an increase in end-diastolic volume (see Figure). If the ventricle now contracts at this increased preload, and the afterload and inotropy are held constant, the ventricle empties to the same end-systolic volume, increasing its stroke volume, which is defined as end-diastolic minus end-systolic volume. The increased stroke volume is represented as an increase in the width of the pressure-volume loop. The normal ventricle, therefore, can increase its stroke volume to match physiological increases in venous return. This is not, however, the case for ventricles that are in failure.
Mechanisms
Increased venous return increases the ventricular filling (end-diastolic volume) and therefore preload, which is the initial stretching of the cardiac myocytes before contraction. Myocyte stretching increases the sarcomere length, which causes an increase in force generation and enables the heart to eject the additional venous return, increasing stroke volume.
This phenomenon can be described in mechanical terms by the length-tension and force-velocity relationships for cardiac muscle. Increasing preload increases the active tension developed by the muscle fiber and increases the velocity of fiber shortening at a particular afterload and inotropic state.
One mechanism to explain how preload influences contractile force is that increasing the sarcomere length increases troponin C calcium sensitivity, which increases the rate of cross-bridge attachment and detachment, and the amount of tension developed by the muscle fiber (see Excitation-Contraction Coupling). Other mechanisms are undoubtedly involved. The effect of increased sarcomere length on the contractile proteins is termed length-dependent activation.
It was taught for several decades after Starling's work that the Frank-Starling mechanism results from changes in the number of overlapping actin and myosin units within the sarcomere, as in skeletal muscle. According to this view, changes in the force of contraction do not result from a change in inotropy. Because we now know that changes in preload are associated with altered calcium handling and troponin C affinity for calcium, a sharp mechanistic distinction cannot be made between length-dependent changes (Frank-Starling mechanism) and length-independent changes (inotropic mechanisms) in contractile function.
Revised 01/23/2023