 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Renin-Angiotensin-Aldosterone System
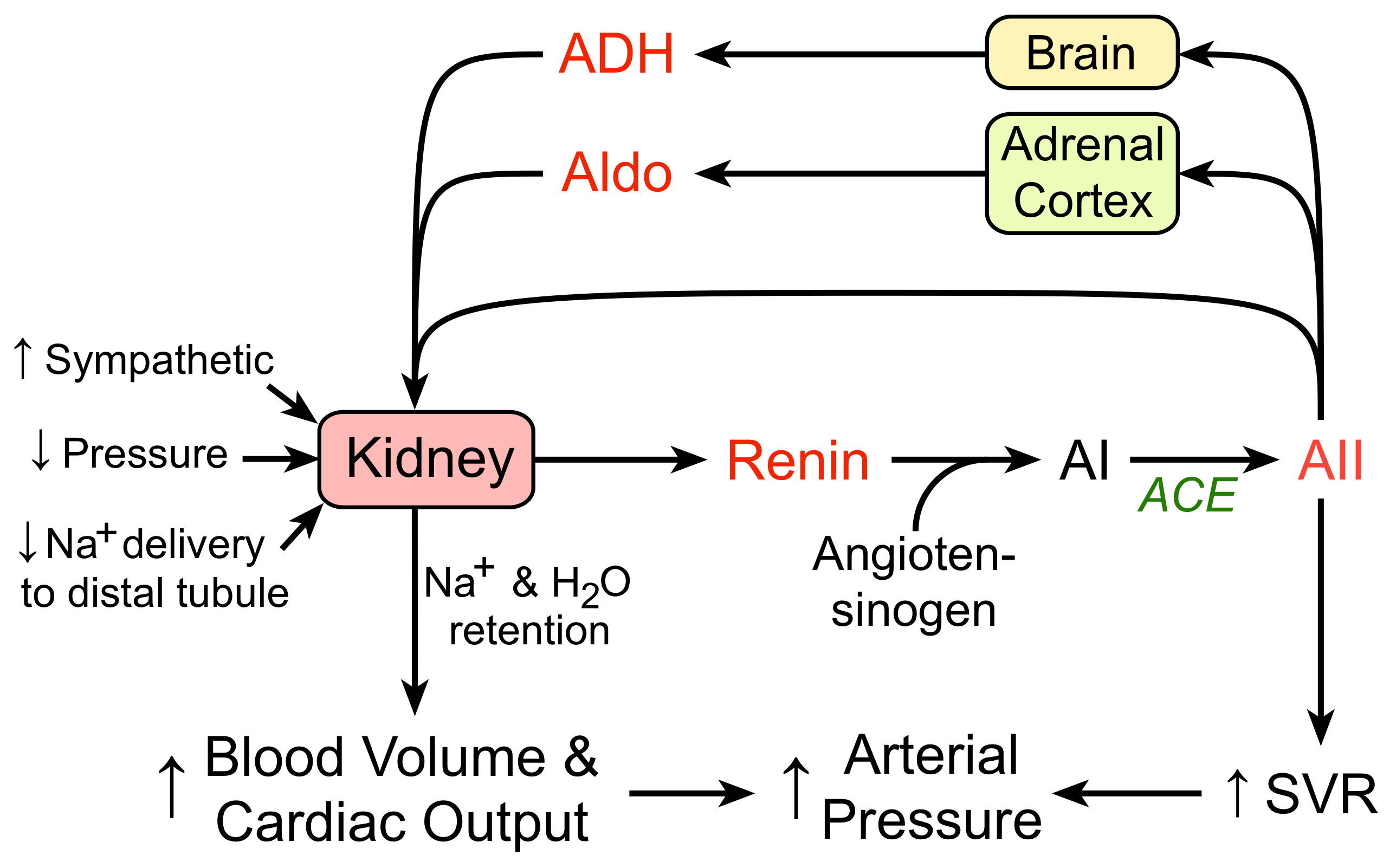
The renin-angiotensin-aldosterone system (RAAS) plays an important role in regulating blood volume and systemic vascular resistance, which together influence cardiac output and arterial pressure. As the name implies, there are three important components to this system: 1) renin, 2) angiotensin, and 3) aldosterone. Renin, which is released primarily by the kidneys, stimulates the formation of angiotensin in blood and tissues, which stimulates the release of aldosterone from the adrenal cortex.
Renin is a proteolytic enzyme that is released into the circulation by the kidneys. Its release is stimulated by:
- sympathetic nerve activation (acting through β1-adrenoceptors)
- renal artery hypotension (caused by systemic hypotension or renal artery stenosis)
- decreased sodium delivery to the distal tubules of the kidney
 Juxtaglomerular (JG) cells associated with the afferent arteriole entering the renal glomerulus are the primary site of renin storage and release. A reduction in afferent arteriole pressure causes the release of renin from the JG cells, whereas increased pressure inhibits renin release. Beta1-adrenoceptors on the JG cells respond to sympathetic nerve stimulation by releasing renin. Specialized cells (macula densa) of distal tubules lie adjacent to the JG cells of the afferent arteriole. The macula densa senses the concentration of sodium and chloride ions in the tubular fluid. When NaCl is elevated in the tubular fluid, renin release is inhibited. In contrast, a reduction in tubular NaCl stimulates renin release by the JG cells. There is evidence that prostaglandins (PGE2 and PGI2) stimulate renin release in response to reduced NaCl transport across the macula densa. When afferent arteriole pressure is reduced, glomerular filtration decreases, and this reduces NaCl in the distal tubule. This serves as an important mechanism contributing to the release of renin when there is afferent arteriole hypotension, which can be caused by systemic hypotension or narrowing (stenosis) of the renal artery that supplies blood flow to the kidney.
Juxtaglomerular (JG) cells associated with the afferent arteriole entering the renal glomerulus are the primary site of renin storage and release. A reduction in afferent arteriole pressure causes the release of renin from the JG cells, whereas increased pressure inhibits renin release. Beta1-adrenoceptors on the JG cells respond to sympathetic nerve stimulation by releasing renin. Specialized cells (macula densa) of distal tubules lie adjacent to the JG cells of the afferent arteriole. The macula densa senses the concentration of sodium and chloride ions in the tubular fluid. When NaCl is elevated in the tubular fluid, renin release is inhibited. In contrast, a reduction in tubular NaCl stimulates renin release by the JG cells. There is evidence that prostaglandins (PGE2 and PGI2) stimulate renin release in response to reduced NaCl transport across the macula densa. When afferent arteriole pressure is reduced, glomerular filtration decreases, and this reduces NaCl in the distal tubule. This serves as an important mechanism contributing to the release of renin when there is afferent arteriole hypotension, which can be caused by systemic hypotension or narrowing (stenosis) of the renal artery that supplies blood flow to the kidney.
When renin is released into the blood, it acts upon a circulating substrate produced by the liver, angiotensinogen, that undergoes proteolytic cleavage to form the decapeptide angiotensin I. Vascular endothelium, particularly in the lungs, has an enzyme, angiotensin converting enzyme (ACE), that cleaves off two amino acids to form the octapeptide, angiotensin II (AII). Note that many other tissues in the body (heart, brain, vascular) also can form AII.
Angiotensin II has several important functions:
- Constricts resistance vessels (via AII [AT1] receptors) and increases systemic vascular resistance and arterial pressure
- Stimulates sodium transport (reabsorption) at several renal tubular sites, increasing sodium and water retention by the body
- Acts on the adrenal cortex to release aldosterone, which acts on the kidneys to increase sodium and fluid retention
- Stimulates the release of vasopressin (antidiuretic hormone, ADH) from the posterior pituitary, which increases water retention by the kidneys
- Stimulates thirst centers within the brain
- Facilitates norepinephrine release from sympathetic nerve endings and inhibits norepinephrine re-uptake by nerve endings, enhancing sympathetic adrenergic function
- Stimulates cardiac hypertrophy and vascular hypertrophy
The renin-angiotensin-aldosterone pathway is not only regulated by the mechanisms that stimulate renin release, but it is also modulated by natriuretic peptides released by the heart. These natriuretic peptides act as an important counter-regulatory system.
Therapeutic manipulation of this pathway is important in treating hypertension and heart failure. ACE inhibitors, AII receptor blockers and aldosterone receptor blockers, for example, are used to decrease arterial pressure, ventricular afterload, blood volume and hence ventricular preload, as well as inhibit and reverse cardiac and vascular hypertrophy.
Revised 12/13/2022