 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Non-Pacemaker Action Potentials
 Atrial myocytes and ventricular myocytes are examples of non-pacemaker action potentials in the heart. Because these action potentials undergo very rapid depolarization, they are sometimes referred to as “fast response” action potentials. (Purkinje cells are fast response action potentials, but possess slow pacemaker activity during phase 4.)
Atrial myocytes and ventricular myocytes are examples of non-pacemaker action potentials in the heart. Because these action potentials undergo very rapid depolarization, they are sometimes referred to as “fast response” action potentials. (Purkinje cells are fast response action potentials, but possess slow pacemaker activity during phase 4.)
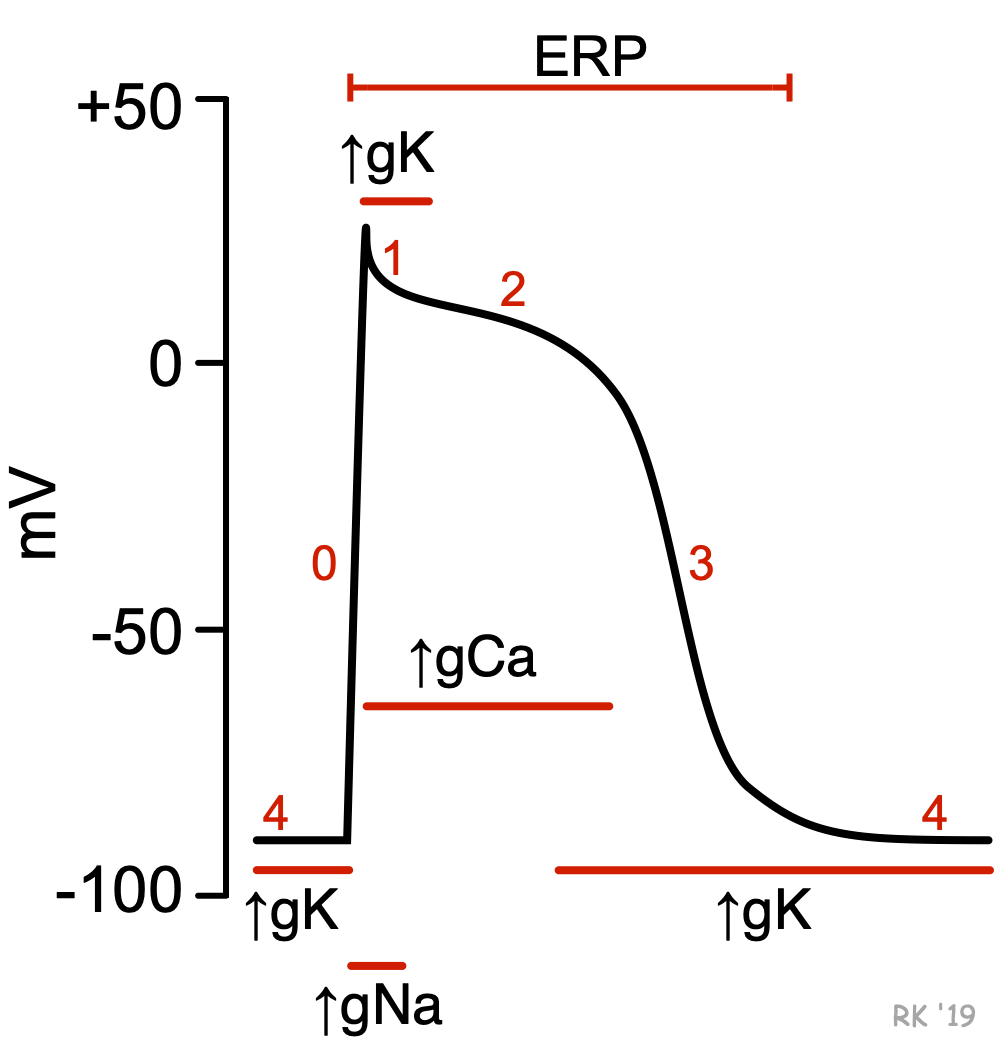
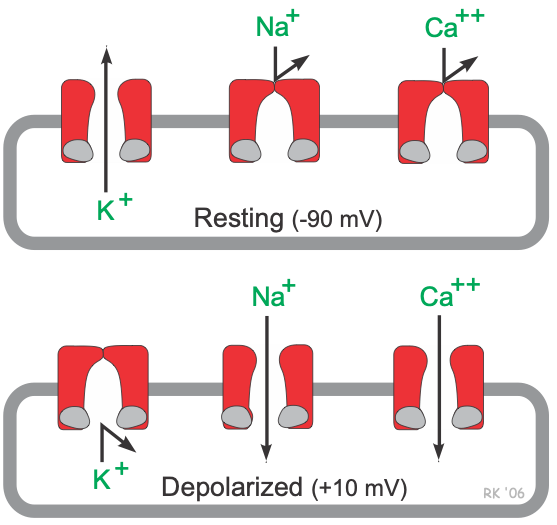
Unlike pacemaker cells found in nodal tissue within the heart, non-pacemaker cells have a true resting membrane potential (phase 4) that remains near the equilibrium potential for K+ (EK). The resting membrane potential is very negative during phase 4 (about -90 mV) because potassium channels are open (K+ conductance [gK] and K+ currents are high). As shown in the figure, phase 4 is associated with increased gK, which causes outward-directed K+ currents. In other words, positive potassium ions are leaving the cell and making the membrane potential more negative inside. At the same time, fast sodium channels and (L-type) slow calcium channels are closed, so Na+ and Ca++ inward currents are very low.
When these cells are rapidly depolarized to a threshold voltage of about -70 mV by an action potential in an adjacent cell, this triggers a rapid depolarization (phase 0). This is caused by a transient increase in fast Na+-channel conductance (gNa) through fast sodium channels. This increases the inward directed, depolarizing Na+ currents (INa) that are responsible for generating the “fast-response” action potentials (see above figure). At the same time, sodium channels open, gK and outward directed K+ currents fall as potassium channels close. These two conductance changes move the membrane potential away from EK (which is negative) and closer toward the equilibrium potential for sodium (ENa), which is positive.
 Phase 1 represents an initial repolarization that is caused by the opening of a special type of transient outward K+ channel (Kto), which increases gK and causes a short-lived, hyperpolarizing outward K+ current (IKto). However, because of the large increase in slow inward gCa occurring at the same time and the transient nature of IKto, the repolarization is delayed and there is a plateau phase in the action potential (phase 2). This inward calcium movement ICa(L) is through long-lasting (L-type) calcium channels that open up when the membrane potential depolarizes to about -40 mV. This plateau phase prolongs the action potential duration and distinguishes cardiac action potentials from the much shorter action potentials found in nerves and skeletal muscle. Repolarization (phase 3) occurs when gK (and therefore IKr) increases, along with the inactivation of Ca++ channels (decreased gCa).
Phase 1 represents an initial repolarization that is caused by the opening of a special type of transient outward K+ channel (Kto), which increases gK and causes a short-lived, hyperpolarizing outward K+ current (IKto). However, because of the large increase in slow inward gCa occurring at the same time and the transient nature of IKto, the repolarization is delayed and there is a plateau phase in the action potential (phase 2). This inward calcium movement ICa(L) is through long-lasting (L-type) calcium channels that open up when the membrane potential depolarizes to about -40 mV. This plateau phase prolongs the action potential duration and distinguishes cardiac action potentials from the much shorter action potentials found in nerves and skeletal muscle. Repolarization (phase 3) occurs when gK (and therefore IKr) increases, along with the inactivation of Ca++ channels (decreased gCa).
Therefore, action potentials in non-pacemaker cells are primarily determined by relative changes in fast Na+, slow Ca++ and K+ conductances and the corresponding ion currents. As described under the discussion on membrane potentials and summarized in the following Goldman-Hodgkin-Katz equation shown below, the membrane potential (Em) is determined by the relative conductances of the major ions distributed across the cell membrane. When g'K is high and g'Na and g'Ca are low (phases 3 and 4), the membrane potential will be more negative (resting state in the figure). When g'K is low and g'Na and/or g'Ca are high, the membrane potential will be more positive (phases 0, 1 and 2) (depolarized state in the figure).
Em = g'K+ (−96 mV) + g'Na+ (+50 mV) + g'Ca++ (+134 mV)
These fast-response action potentials in non-nodal tissue are altered by antiarrhythmic drugs that block specific ion channels. Sodium-channel blockers such as quinidine inactivate fast-sodium channels and reduce the rate of depolarization (decrease the slope of phase 0). Calcium-channel blockers such as verapamil and diltiazem affect the plateau phase (phase 2) of the action potential. Potassium-channel blockers delay repolarization (phase 3) by blocking the potassium channels that are responsible for this phase.
Effective Refractory Period
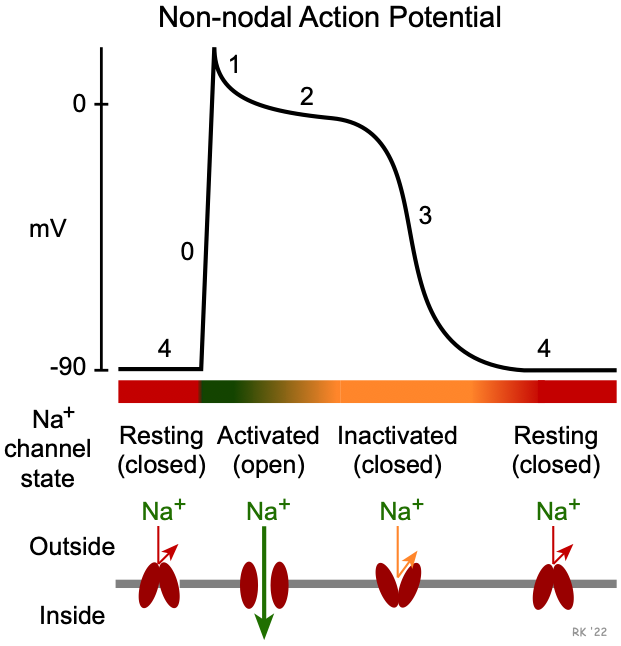 Once an action potential is initiated, there is a period of time comprising phases 0, 1, 2, 3 and early phase 4 that a new action potential cannot be triggered (see figure at top of page). This is termed the effective refractory period (ERP) of the cell. During the ERP, stimulation of the cell by an adjacent cell undergoing depolarization does not produce new, propagated action potentials. This occurs because fast sodium channels remain inactivated following channel closing during phase 1 (see figure). They do not change to their closed, resting (excitable) state until sometime after the membrane potential has fully repolarized. The ERP acts as a protective mechanism in the heart by preventing multiple, compounded action potentials from occurring (i.e., it limits the frequency of depolarization and, therefore, heart rate). This is important because at very high heart rates, the heart cannot fill adequately with blood and therefore ventricular ejection can decrease.
Once an action potential is initiated, there is a period of time comprising phases 0, 1, 2, 3 and early phase 4 that a new action potential cannot be triggered (see figure at top of page). This is termed the effective refractory period (ERP) of the cell. During the ERP, stimulation of the cell by an adjacent cell undergoing depolarization does not produce new, propagated action potentials. This occurs because fast sodium channels remain inactivated following channel closing during phase 1 (see figure). They do not change to their closed, resting (excitable) state until sometime after the membrane potential has fully repolarized. The ERP acts as a protective mechanism in the heart by preventing multiple, compounded action potentials from occurring (i.e., it limits the frequency of depolarization and, therefore, heart rate). This is important because at very high heart rates, the heart cannot fill adequately with blood and therefore ventricular ejection can decrease.
Many antiarrhythmic drugs alter the ERP, thereby altering cellular excitability. For example, drugs that block potassium channels (e.g., amiodarone, a Class III antiarrhythmic) delay phase 3 repolarization and increase the ERP. Drugs that increase the ERP can be effective in abolishing reentry currents that lead to tachyarrhythmias.
Transformation of non-pacemaker into pacemaker cells
It is noteworthy that non-pacemaker action potentials can change into pacemaker cells under certain conditions. For example, if a cell becomes hypoxic, the membrane depolarizes, which closes fast Na+ channels. At a membrane potential of about –50 mV, all the fast Na+ channels are inactivated. When this occurs, action potentials can still be elicited; however, the inward current is carried by Ca++ (slow inward channels) exclusively. These action potentials resemble those found in pacemaker cells in the SA node, and can sometimes display spontaneous depolarization and automaticity. This mechanism may serve as the electrophysiological mechanism behind certain types of ectopic beats and arrhythmias, particularly in ischemic heart disease and following myocardial infarction.
Revised 10/27/2023